|
|
Molecular Modeling Pro Plus On-line Help (v. 9) |
|
Draw
Molecules | Optimize Geometry | Create Databases | Edit
Databases | Calculate Properties |
|
|
Quantum Mechanics | Automated Data Analysis | Atom Based Analysis| Analyze data Manually (tutorial) | Graph data | |
|
|
The Drawing Window - menu item descriptions
| The Database Window - menu item
descriptions |
|

This web page is a basic
introduction to the program. More
complete descriptions of the program and tutorials can be found in the printed
manual and the item by item descriptions of the Drawing Window
menus and the Database Window
menus.
Two tips on installation
1. The first time you use the
program you will want to initialize the links to companion programs. Below are some default settings (File
menu/Initialize Links). If you install the
program in some directory other than c:\molsuite\mmp then the filenames below
should be changed to reflect this. MMP+
should do this automatically. If you do
not have ChemSite then that text box should be left blank. If you have a Brookhaven pdb file viewer put
its filename in the Protein data base viewer box below (Rasmol, found as
freeware on the internet is an example).
If you check Open in database mode and give a database file name, that
database will open every time you open MMP+.
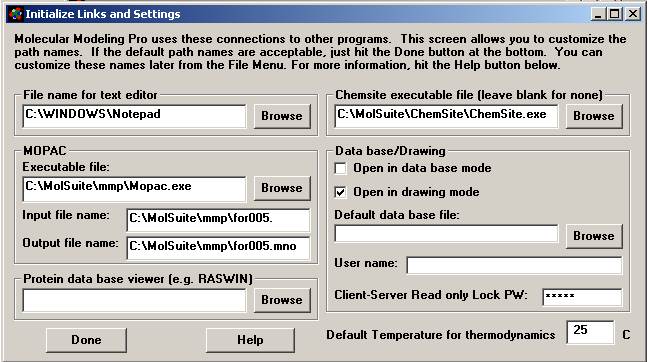
Note: Your path name will not
be c:\molsuite\mmp if you installed the program in a different directory.
Drawing molecules is easy
with this program. Before starting note
that a blue message box often appears near the top of the screen which contains
information on the status of the drawing window and which will help guide you
through the drawing process.
- The first atom: Select the atom type if other than carbon from the buttons at
the left of the screen (select other if not H, C, H, N, O, F, P, S, Cl,
Br, or I). Click on the drawing
screen and the atomic symbol for the atom appears on the screen.
- Subsequent atoms: If the next atom is not the same atom type,
select the atom type again. Select the bond type (the 1, 2 or 3 button
near the left of the screen) if other than a single bond. Click on the atomic symbol at the screen
center. The diatomic molecule is
drawn as a line with the atoms at either end of the line. Repeat this step as desired to draw the
molecule. Click on the atom you
wish to add to after selecting the atom type and bond type. If you are not changing the atom type
and bond type then you do not have to select them. Bond lengths and angles are drawn using
correct values from the literature.
Molecules are drawn 3-dimensional, not 2-dimensional. If the molecule goes off the edge of the
screen use the "Center" button near the top of the screen to
center the molecule. If because of
the 3-D nature of the molecule the atom you wish to add to is difficult to
click on, rotate the molecule by holding down the right mouse button. Holding the mouse button down at the top
or bottom of the screen rotates the molecule along the x axis and holding
it down near the left or right sides of the screen rotates the molecule
around the y axis. You can speed up
the rotations by hitting the "<" and ">" buttons
which appear near the upper left of the screen while rotating.
- Adding rings: Most of the rings commonly
found in organic chemistry are in the ring library that comes with the
program and the list of these rings is accessed through the Ring button
found at the left of the screen.
Select the ring from the list and connect to the previously drawn
atoms by clicking on the atom in the ring and the atom previously drawn
that should be connected.
Some rings are not in the library of course. You have three options for drawing these
rings. (a) select a ring from the
library list and change it, (2) type in the number of atoms in the ring and
have the generic ring builder create a ring which you will have to change where
atom types and bond types are not what you want (also accessed through the Ring
button) or (3) draw the atoms in the ring one by one as above and close the
ring using the "Connect" button near the upper right of the
screen. Before connecting the atoms you
will probably need to rotate some torsional bonds (Rotate menu/bonds). With items 2 and 3 you will need to Minimize the molecule to get good ring geometry.
- Making changes to atoms previously drawn: To
change the atom type click on the Change button at the left, then the atom
type then on the atom to be changed.
To change the bond type click on the Change button, then the 1, 2
or 3 button and then click on the two atoms which are bonded. To delete an atom, click on the Delete
button, then on the atom to be deleted.
The delete and change modes stay on after you change an atom, so if
you want to add an atom after deleting make sure you hit the "Add
button". To delete a bond hit
the "0" button at the left and then click on the two bonded
atoms.
- Tips: Clicking on the H (hydrogen) button
twice is a quick way to add hydrogens to all unfilled valences. Clicking on the Delete button twice
brings up a dialog that allows you to delete whole molecules. If you are not sure what is going to
happen when you click on the screen look at the blue box.
- Tip 2:
The Periodic Table that appears when you hit the "Other" button
at screen left is linked to atomic properties. Bring up the table then Right mouse
click on an atomic symbol to bring up its properties.
Step by step: Drawing benzamide and water together on
the screen.
- If you have a molecule drawn clear it by clicking
the Clear button in the upper right part of the screen.
- Click on the Rings button near the screen left.
- Select benzene from the list.
- Click on the "1" button and the
"C" button near screen left (if you have just turned the program
on you don't need to do this as these are the defaults).
- Click on one of the carbons on the benzene
ring. You have now drawn toluene.
- Click on the "2" button and the
"O" button. Click on the
carbon not on the ring. You have
benzaldehyde.
- Click on the "1" button and the
"N" button. Click on the
carbon not on the ring. You have
benzamide.
- Click on the "H" button twice. Select "Yes" when asked if you
want to add hydrogens to all atoms. (figure 1)
- Rotate the molecule. Hold the right mouse button down on the
drawing screen. Move the mouse
arrow around and note the effect.
Center the molecule on the screen using the "Center"
button.
- Let's display the molecule as shaded
spheres. Go to the Display
menu. Choose Change Display Mode from the menu. Check the Space Filling box. Hit
Done. (figure 2)
|
|
|
|
Figure 1 |
Figure 2 |
- You can also view the molecule in a separate window with much better graphics capabilities. In the upper left of the window, next to the Clear button is a little picture of a molecule. Click on this. Rotate the molecule in the new window by hold down the right mouse button. Translate the molecule by dragging with the left mouse button. Make the molecule larger or small with the page up and page down keys. Change the display mode using the Display menu.
- Saving the molecule: Go to the File menu. Choose Save Molecule from the menu.
Select MDL Molfile from the Save
as Type list. Select the
directory to save the molecule to.
Type in benzamide or benzamide.mol and hit the Save button. Opening the file the next time you use
the program is similar but uses the Open
Molecule menu item instead. MMP
Plus supports the following file types: MDL Molfile, Macromodel,
Brookhaven pdb, MOPAC input and output files, CML (chemical mark-up
language). It fully supports input
of Brookhaven pdb files but only supports the output of these files using
the atom type and bond type descriptors (losing the residue and other
information). You can also input
and output MDL Molfiles over the clipboard (Edit menu/Copy) and output
SMILES notation (Calculate menu).
- Add a second molecule to the screen. Hit the "New Mol" button near
the top center of the screen.
Select the "O" button.
Click anywhere on the drawing screen. Hit the H button twice and select
Yes. You have drawn water.
Minimizing energy to get good 3-D
geometry:
The geometry minimizers are
accessed from the Geometry menu. Choose the menu item :Minimize."
|
|
Use of the the Minimize Geometry window Full MM2 is the standard geometry minimization
procedure. It finds local minima. If you need to have sterochemistry
preserved AND find global minima, run conformational analyses (first menu
item on the Geometry menu) before running MM2. Quick MM2 finds global minima but may ruin stereochemistry If these methods don't work
we recommend the Use Simplex minimization method. This method is slow and sure. Sometimes combining it with MM2 iteratively
gives the best answers. The Moly
minimizer "Refine" routine may be a final resort if all else
fails but is the least recommended method. If you own the program ChemSite, the Amber force field
and minimizer found there is a standard minimizer for macromolecules. |
|
Figure 3 |
|
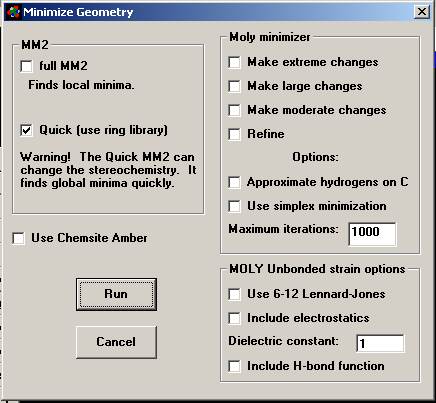
The
standard minimizer in the program is a version of MM2 (Molecular Mechanics)
developed by Norman Allinger and co-workers.
The version here was programmed by P. Baracic and M. Mackov and has an
additional feature - the "Quick" minimizer. Use the "Full MM2" when you
want the stereochemistry to be preserved
and only desire a "local minimum" (e.g. the lowest energy model in
the current conformation). The Quick
minimizer finds a global minimum energy conformation by first taking the
molecule apart based on the location of torsional angles, then minimizes energy
of the parts, then reattaches the bonds with a conformational analysis around
the torsional bonds. In the process the
Quick minimizer uses a ring library to speed up the operation. The Quick minimizer can destroy the
stereochemistry that you carefully have built in the molecule, so keep this in
mind. Otherwise it is more powerful than
the "full MM2" despite its name.
The MM2 minimizer looks up optimum values for
bond lengths, bond angles and torsional bond angles. If an atom combination is missing from the
database it returns an error. In this
case you may have to go to the Edit menu and choose Undo from the menu to
retrieve your molecule. Molecules of
greater than 999 atoms cannot be run through the MM2 minimizer. Atom combinations not typically found in
organic chemistry should probably not be run through MM2 either. If you have some of these exceptions, use the
MOLY Simplex minimizer.
With
the MOLY and Simplex minimizers you can see the minimization take place on the
screen. With the MM2 routine you just wait
and the minimized molecule is eventually returned from the MM2 DLL.
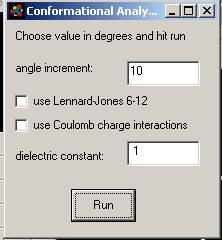
Conformational Analysis
|
|
Conformational Analysis is
the first item on the Geometry menu.
It allows you to rotate torsional bonds while calculating strain
energy due to atom atom overlap, Coulomb charge interactions and hydrogen
bonding. In the example below (figure
5) clicking on carbon atoms 1 and 2 rotates the bond shown so that the
isopropyl group rotates around the axis formed by the bond. You can rotate two bonds at once. At the end of the analysis the program sets
the molecule in the minimum conformational geometry and prints out the chart
in figure 6. Using the conformational
energy routine iteratively may be necessary if you have more than 2
conformationally flexible bonds. You
also may often want to run this routine before the MM2 "full"
minimizer in order to obtain global energy minima (e.g. the best possible
geometry). |
|
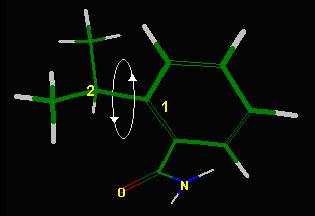
Figure 4 Figure 5 --> |
|
|
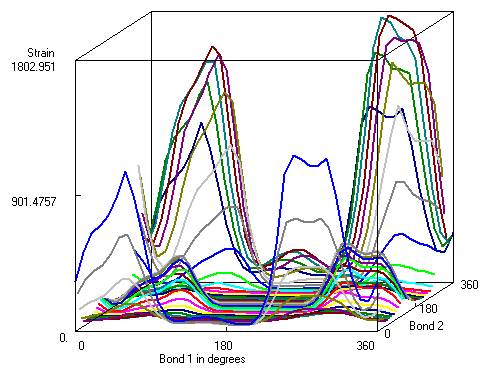
Figure 6 --> Conformational analysis of the molecule shown in
figure 5. Bond 1 is rotation of the isopropyl
group and bond 2 rotation of the amide group (the aryl-C bond forms the axis
of rotation). Strain energy is in
kcal/mole with lowest energy the best conformation. |
|
MOPAC
Geometry Optimization
Perhaps
the best geometry optimization method is using the PM3 force field in
MOPAC. The caveat being it too only
finds a local minimum. See the section
under Quantum
Mechanics for more details.
Geometry
Optimization of whole directories of molecules
You can optimize whole directories of molecules from
the menu item under the Geometry menu entitled minimize all molecules in a directory. The process is similar to that described
above for single molecules except you must additionally tell the program which
directory to use. Molecules must be
stored in Macromodel or MDL Molfile formats.
Any of the minimizers can be used (MM2, Simplex, MOLY, ChemSite
Amber). The MM2 Quick minimizer was
especially designed to turn 2-D molecules into 3-D molecules. Remember, though, that it changes
sterochemistry, so take some care with 3-D molecules where sterochemistry is
important.
Adding
a molecule to a database is as simple as: a) select Save Database from the File menu of the drawing window. and Save this molecule to database from
the submenu; b) select the directory and type in the database name; c) if the
structure is saved in a separate file (this is an option described later) type
in the structural file name and the program will do the rest. However, if the database is not yet made
there are some additional things to do which are described next.
The
first step in creating a new database is the same as adding a molecule to an
existing one:
·
Select Save
Database from the File menu and Save this molecule to Database from the submenu
·
Select the
directory. Select the type of database
from the list at the bottom. The MAP
.csv file type is no longer recommended unless you are using the old program
Molecular Analysis Pro (it is limited to 72 numeric fields and MMP+ is capable
of creating databases with many times more fields). Microsoft ACCESS or XML are both fine for
creating databases. If you are going to
store reactions then use ACCESS Type in the database name. There is an incompatibility with the version
of ACCESS used by MMP and Windows 10.
·
The next window
looks much like a file saving window, but consider carefully if you want to store
the structures within the database itself or as separate connection tables
(e.g. MDL Molfiles, Macromodel files, Brookhaven pdb files etc.). Storing them internally will create one very,
very large file if you have a large database.
Storing them in connection table files will create potentially thousands
of small files in a standard file format such as the MDL Molfile.
·
Next to appear is
the window where you select which properties to store (figure 7).
|
|
Figure
7.
Property Selection Window after the Check All fast fields box has been checked. |
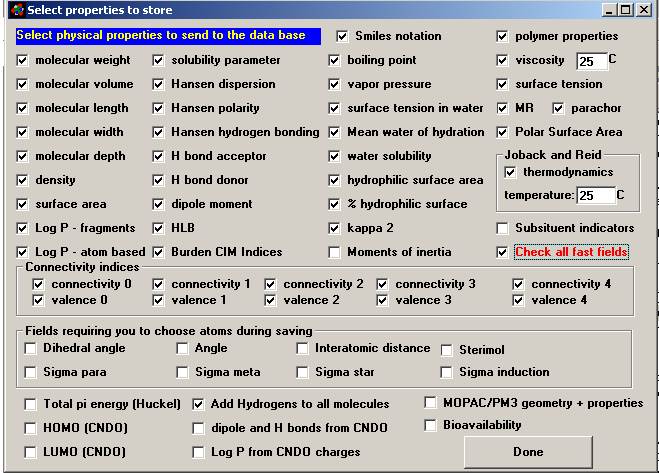
In the
example above the Check all fast fields (ones which are quickly calculated) was
checked which caused the majority of the fields on the screen to also be
checked.
If
you check any of the "Fields requiring you to choose atoms during
saving" then if you later wish to add whole directories of molecules to
the databases you will have to sit by the computer and select atoms in each
molecule for the properties you select.
For instance if you select interatomic distance you will have to select
the two atoms involved. These properties
can be useful in QSAR studies, they will just slow down batch database
creation.
The fields
at the bottom of the screen with the exception of Bioavailability require CNDO
or MOPAC calculations and can take a minute or two to complete. Including these fields in a large database
will add hours to the time it takes to calculate the properties. However, the properties calculated can be
very useful.
Some
of the property boxes select multiple properties and place multiple fields in
the database. These include the Burden
CIM indices, Bioavailability, MOPAC/PM3 geometry + properties (which also does
a geometry optimization using MOPAC - which will be stored), polymer properties
and the Joback and Reid thermodynamics.
It especially includes the substituent indicators which will add over
160 fields (substructure fragments like the number of carbons, number of ester
groups, number of amides etc.) to the database.
See
the Calculate
Properties section below for more
details on physical property calculations.
·
The program will
ask you to type in a name for the molecule.
This field cannot be blank.
·
The database will
then be created and you will be asked if you want to see it. You should take a look and make sure that the
program has done what you wanted it to do.
Besides adding molecules from the File menu of the
drawing window, you can also add molecules to the databases from the Edit menu
of the Database window. See The Database Window -
menu item descriptions.
Tip: A database of industrial chemicals called
industrial.mdb is installed with MMP+.
This file contains over 800 molecules and can be the starting point for
creation of a large database of commonly used chemicals. It also contains information about the
compounds from the literature that you may find useful.
Creating databases from SMILES notation
If you know how to write SMILES structures a short-cut
method of creating a database from scratch is detailed in this section:
1. Open Excel. In
the first row, write Name in column 1 and SMILES in column two. Input the names and SMIILES strings in the
two columns below these headers.
2. You may also put experimental data (with a field name
in row 1) in the EXCEL spreadsheet.
3. Save as a Windows csv file.
4. Open the file in MMP+ (File/Open Database). Select .csv file as the file type. Select the file name.
5. The following window will appear.

Checking
items will case MMP+ to calculate properties of all the molecules and add them
to the database. MMP+ will automatically
calculate 3-D structures, molecular formulas and molecular weight even if you
check nothing. It will look in the file
models\CASNumbers.csv for a name match and if found, will add CAS numbers to
the database.
Remember to Save the database if you
want to keep the changes.
Creating
databases in batch mode
Besides saving molecules one at a time, there are ways
to create databases or add molecules to existing databases many at a time. If molecules are stored in a directory in one
of the standard connection table formats that the program reads (e.g. MDL
Molfiles, MACROMODEL files), then you can use the batch procedure in MMP+ that
will take all the molecules in the directory and add them to the database. The procedure is almost identical to saving
an individual molecule. Go to the File
menu of the drawing window and select Save
Database/All Macromodel files in a directory or Save Database/All Molfiles in a directory.
Creating
a QSAR database of closely related analogs from a substructure
Draw in the substructure. Select from the File menu of the Drawing
window - Save Database/Make QSAR database... You then select one, two or three sites on
the molecule to vary and tell the program what the database name should be and
where you want to store it. You can add
the molecules to an existing database or create a new one. The program automatically will add the various
substituents to the parent structure previously drawn. If you only vary one site it adds all the
substituents listed in the file aromqsar.txt (aromatic substituents) or
alipqsar.txt (aliphatic subsituents). If
you vary two or three sites then it adds “x” squared or “x” cubed molecules to
the database (uses the first “x” substituents in the files aromqsar.txt or
alipqsar.txt where “x” is a number you choose.). If you edit the text files be sure to save
them first to a backup file (you can add substituents by editing these files if
you have literature values for the fields in the files and have saved a
connection table). The physical property
fields created by the routine are somewhat different than those created by
other routines in MMP+. Many of the values
are substituent values from the literature (pi, mr, sterimol parameters, sigma)
and are obtained from the tables in the two text files.
Note that modifying three sites can quickly create a
very large database. For instance,
setting “x” to 30 would result in 30x30x30 = 27000 molecules. At present keep three substituent databases
to “x”=31 or less (the program contains 16 bit integers with 32000 the upper
limit.)
Step by
Step - Creating a QSAR database
·
Draw
benzamide. If you need instructions, go here...
·
Go to the File
menu. Select Database Save and Make QSAR database
·
Click on the
hydrogen para to the amide group.
·
We will only vary
one substituent site, so hit "No" when it asks if you want to vary
another.
·
Name the database
HelpExample and save it to the /mmp root directory.
·
Although it will
take a while, let's have the program minimize with MOPAC/PM3 and save some of
the quantum mechanics properties (check the third check box in figure 8
below). We are going to create one
indicator variable two so change the text box displaying a zero to a
"1". The QSAR database
creation adds the substituents with a crude (30 degree) conformational
analysis. MOPAC PM3 will take care of
the fine geometry minimization.
|
|
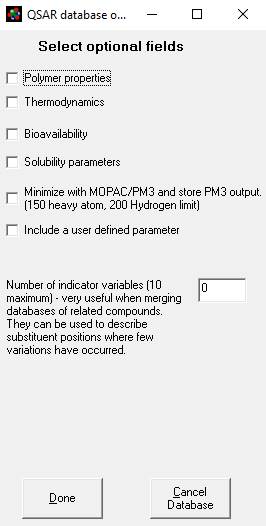
Figure
8. The QSAR database creation options
screen. Each check box will save
several additional calculated fields to the database. Checking polymer properties will save a
number of properties using van Krevelen's methods. Thermodynamics saves the Joback and Reid
method calculations. Bioavailability
saves the Rule of 5 calculations (0 = uptake likely), Log P (Moriguchi
method) and the number of hydrogen bond acceptors and donors. Solubility
parameters save a number of properties associated with solvation, like the
Abraham solvation parameters and surfactant properties like HLB. User defined
properties are the ones created from the Database Window’s calculate menu. Indicator variables are useful when running
this routine iteratively (e.g. using different substructure templates and
merging the data). The indicator
variable values will distinguish each iterative run from the next. |
·
The first three letters
of the Macromodel file names we will set to bap (for benzamide and para).
·
The building
block structure we will call benzamide (p-)
·
The value for
indicator 1 we will set to "1".
·
The database will
now be created. Most of the time spent
will be in the MOPAC calculations. If
the program asks if you would like to add some hydrogens, answer
"No".
·
When the program
asks if you would like to open the database answer "No".
We now will repeat the process with meta substitution:
·
Go to the File
menu. Select Database Save and Make QSAR database
·
Click on the
hydrogen
·
We will only vary
one substituent site, so hit "No" when it asks if you want to vary
another.
·
Name the database
HelpExample and save it to the /mmp root directory.
·
When the program
asks if you want to overwrite the database select "No".
·
When the program
asks for the three letters of the Macromodel file names set them to bam (for
benzamide and meta).
·
The building block
structure we will call benzamide (m-)
·
The value for
indicator 1 we will set to "0".
·
The database will
now be created. Most of the time spent
will be in the MOPAC calculations. If
the program asks if you would like to add some hydrogens, answer "No".
·
When the program
asks if you would like to open the database answer "Yes".
·
In the database
window go to the View menu and select "File Card". Your screen should be displaying something
similar to figure 9 below.
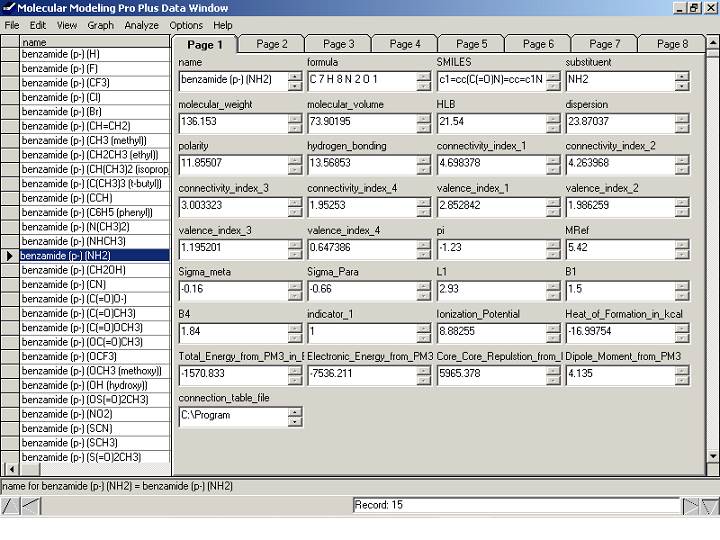
Figure 9. QSAR database created by our step by step
tutorial. The following values are for
the substituent (not the molecule as a whole) and are literature values from
tables: pi (lipophilicity), MRef (molar refractivity), Sigma_meta, Sigma_para,
L1 (length), B1 (minimum width), B4 (maximum width). Indiacator 1 is "1" for para
substitution and "0" for meta substitution. The fields after indicator_1 were calculated
by MOPAC. The values before pi are
molecular properties calculated by MMP+.
Tip: clicking on the gray area near the bottom ["name for benzamide
(p-)(NH2) = benzamide (p-)(NH2)] will bring it full screen. Clicking again will bring back the above
view. This allows you to view overly
long text fields.
To save changes to the
databases you must go to the File/Save menu and tell the program to save the
data. Desktop databases are by default
backed up every time you open one (to database filename.bak).
Basic Editing tasks
To change values simply type
into the spreadsheet cells. When you
have made the changes save the data (File menu).
Tip: Memo fields can contain
over a megabyte of data. Memo fields
include and field named "Name", SMILES, formula, general_information, trade_name,
safety_data, and IUPAC_name. To view all
the information in one of these fields click on the gray area near the bottom
of the screen containing the text information.
This will bring it full screen (you cannot edit the information here
however). To bring back the spreadsheet
view simply click on the text again.
Definitions: Record =
molecule; Field = molecular property
To add a new molecule to the
database draw in the molecule, then go to the database window's Edit menu and
choose Records(molecules) and Add Molecule. Follow the prompts.
To delete a molecule, select
the molecule in the database spreadsheet by clicking on it. Go to the Edit menu and choose Records(molecules) and Delete current molecule. Follow the prompts.
Record (molecule) additions
and deletions do not take effect until you save the data.
Reactions
Databases of reactions can be
made as well. These databases require
two tables so must be stored in a database format that uses multiple tables
like Microsoft ACCESS, Microsoft SQL Server or ORACLE. For more information on creating reaction
databases see the description HERE.
Substructure Searches
Load the database. Draw in
the substructure. Go to the Edit
menu. Choose Substructure Search. The program will ask you if you want to create
a dataset of the molecules containing the subset. Usually you will want to choose NO here,
especially if you have made changes to the database and not saved them. Choosing YES will create a new database
containing only the molecules containing the substructure (which you can make
permanent by saving). Choosing NO will
give you a list of molecules which contain the substructure from which you can
select molecules in the current database.
For more go HERE.
For a more complete
description of the menu items in the database window go HERE.
Calculation of
Chemical Properties from Structure
Physical properties are
calculated from structure using methods garnered from the literature, and by
the MOPAC and CNDO programs, and with methods developed internally by Norgwyn
Montgomery Software Inc. Most of these
properties are accessed from the Calculate menu of the drawing window. They also are calculated during database creation. The basic
procedure is to draw the molecule, then simply select the properties you wish
to calculate from the Calculate menu. MOPAC and CNDO properties are accessed from the Tools menu. A description of the properties calculated
are HERE. The menu
items found under the calculate menu are described in: The Database Window -
menu item descriptions. Figure 10 shows the Calculate menu.
With the advent of version 8 of the program, the old
methods are being replaced by new methods created with the MMP Substructure
Analysis routine which gives superior models of properties. These models are documented and are
downloadable from the COMMUNITY sharing page at https://www.norgwyn.com/models/Models.html.
|
|
Figure 10. The Calculate
menu is used to find calculated values for the molecule drawn. Most of the calculations have limitations
to organic molecules containing H, C, N, O, F, Si, P, S, Cl, Br and I. A description of the properties is found HERE. MOPAC and CNDO physical
property calculations and geometry optimization is done from the Tools menu. Many of the menu items
contain multiple property calculations.
For instance, the Solubility Parameter menu item is linked to
calculations for the solubility parameter (3 methods), HLB, Log octanol water
partition coefficient (Log P), water solubility, mean water of hydration,
polar surface area, % hydrophilic surface area, and the Hansen 3-D solubility
parameters. |

Quantum
Mechanics
Molecular Modeling Pro Plus supports two public domain
semiempirical quantum mechanics programs: CNDO and MOPAC (since version 6 this
is included). The program MOPAC.exe
included with this program was developed at the Air Force Academy and the
version here was adopted for Windows by Victor Lobanov at the
The program CNDO (Complete Neglect of Differential
Overlap) is the earlier of the two programs and was developed by J. Pople and
co-workers in the 1960s and 1970s. It is
still thought one of the best ways to calculate partial atomic charges and
dipole moments. For most other uses,
though, the MOPAC methods are more sophisticated and faster. Pople abandoned semi-empirical methods and
worked on ab-initio calculations after CNDO.
MOPAC was developed by J.J.P. Stewart and colleagues
at the Air Force Academy. It is actually
a collection of methods semi-empirical quantum mechanics methods developed by
them and by M.J.S. Dewar and colleagues at the
|
|
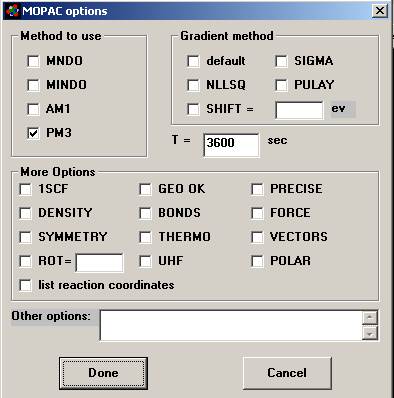
·
Figure 11. Enter the MOPAC screen from the Tools menu
of the drawing window. Choose the
method to use. The AM1 and PM3 force
fields are more modern than the MNDO and MINDO methods. MNDO and PM3 support the most atom
types. If you checke POLAR - the
polarizability and first and
second hyperpolarizabilities are to be
calculated. THERMO - Thermodynamics calculations can be performed on
molecules. The key words FORCE and ROT
also must be included. The combination of these three key words will give the
user additional thermodynamic property calculations: internal energy, heat capacity, partition function and entropy for translation,
rotation and vibrational energy over a range
of temperatures |
If you do more than one iteration (don't check 1SCF),
MOPAC will find a local geometry energy minimum (i.e. will return changed,
optimized atom 3-D coordinates) and MMP+ will display the new geometry. MMP+ will also cause the MOPAC output to be
displayed in a Notepad window. If the
program comes back with the message “No geometry read in for some reason,” go
to Edit and choose Undo. If the starting geometry is bad minimize the structure
(Geometry/Minimize menu.) Rerun MOPAC with GEOOK checked. The limit on molecule size for this version
of MOPAC is 150 non-hydrogen atoms plus 200 hydrogens.
For a more complete description of MOPAC and CNDO see The Drawing Window - menu item descriptions . A description of the MOPAC output is found HERE.
Molecular Modeling Pro Plus has three methods for automated generation
of QSAR/QSPR models which were designed to let chemists with a general
knowledge of statistics create usable models fairly quickly. You access these
methods by first opening a database. You must add the experimental data you
wish to model to the database by typing it in or importing it from Excel. Next
you just select either "Substructure Analysis" or "Stepwise
regression" from the Analyze menu of the Database window. The Substructure
Analysis routine will analyze all the molecules in your database for the unique
sub-structures contained in the structures and make variables from the
molecular fragments to correlate (regress) against the experimental data. The
Stepwise regression model will use all the existing fields (calculated and
experimental properties) already contained in the database instead of
sub-structural fragments. A further explanation follows.
To access this
feature: Open a database. Go to the Analyze menu. Choose the first item,
"Substructure Analysis."
What this
analysis will do: After you choose the property to model, this method will
analyze all the structures in your database in order to create independent
variables of the substructures contained within the molecules. It will then use
least squares multiple regression to find the substructural features that are
significantly correlated with the property and places the model in your \models
subdirectory. After completing the analysis, this property can be predicted for
any molecules you draw by selecting "user defined properties" from
the Calculate menu of the Drawing window.
What can be
analyzed: This method does not care what atom types you have in your database.
It creates the substructures from the molecules themselves, not from a look-up
table. You should be able to analyze any set of molecules as long as you have
input the structures correctly. Molecules should be drawn in a consistent
manner (e.g. if you draw nitro as N(=O)=O one time and N+(=O)O- the next it
will not recognize them as the same substructure. Salts and ionic molecules are
also types of molecules that can be drawn in many different ways.)
CAVEAT EMPTOR:
On large databases this method will take days to complete the task. On a
database of 72 structurally related molecules this method takes a few minutes
to complete its task on a slow XP computer. On a database of 800 molecules
containing mostly solvents and surfactants, this method took 2 hours to
complete its task on a slow XP computer. On a database containing over 4000
molecules with over 1300 unique sub-structural features on an Intel I5, Windows
10 computer the program took about a week to converge. Fortunately, the
computer was still usable as the task ran in the background. However, the
database being used was tied up for the duration of the analysis. The length of
the task is a function of your computing speed. The size of the database that
can be used is limited by RAM.
Details of the
Method:
The number of
variables that will be generated is a function of the size and complexity of
the structures in the database. The default setting for maximum number of
sub-structural variables is 1500. If you get an "out of memory" error
go to the Options menu of the Database window and decrease the number of
variables allowed in the analysis. If using the mydata.mdb or chemelectrica.mdb
database you will need to set this to about 1500 because the program will find
about 1300 substructures (as of January 2018).
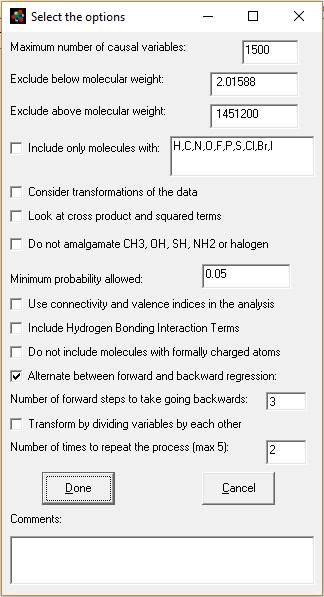
Explanation of this window:
1. Maximum number of causal variables: The program
will calculate this based on the size and complexity of your database, up the
maximum allowed (see previous paragraph). You can adjust this number here and
in the Options menu.
2. Exclude by molecular weight (boxes 2 and 3): You
can limit the analysis to only consider molecules between a minimum and maximum
molecular weight.
3. Include only molecules with: - Check this box if
you want to only want to analyze molecules containing specific atom types. You
can modify which atoms you look for by adding or subtracting atomic symbols
from the comma separated list.
4. Consider transformations of the data:
If checked, the program will try to determine the best
transform for the data (i.e transform the experimental response
parameter.) It will do this using only
for the models of the count for each type of atom as independent variables
(i.e. number of carbons, number of hydrogens etc.). The transforms of response used by MMP are a
combination of: (a) multiply or divide the raw data by molecular weight and/or
molecular volume, and (b) log10, square, square root or logistic and the
inverse of these five transforms. A
total of 70 different combinations of these factors will be analyzed. The program will choose one, then proceed
with the rest of the analysis. If you do
not choose to consider the transformations, the program will still see if the
data is better fit by dividing all the atom counts (e.g. number of carbons,
number of oxygens, etc.) by molecular weight (it will not do this if you select
transformations, as multiplying the response by molecular weight is similar to
dividing the independent variables by molecular weight.)
5. Look at cross product and squared terms. After the model completes the stepwise
regression of all the independent variables, it will look the squares and cross
products of all the terms in the model to see if they improve it further.
6. Do not amalgamate CH3, OH, SH, NH2, NO2 or halogen.
If checked, the method will use a simpler, faster set
of fragments to analyze the data. You would complete the analysis sooner and
perhaps have a more general model, but lose some of the structural information
in the data.
MMP+ combines terminal groups, like H, OH, =O, =S, F,
Cl, Br, I, NH2, #N, NO2 and CH3 with the atoms they are attached to in the
default model. It also looks for concentrations of heteroatoms with structures:
O[or N]C(=O) [or =S]N [orO]. If you check this box the program will only attach
H, =O and =S.
If you check the box the molecule CH3CH2C(=O)NHCH2CH3
would generate the following pairs for analysis:
Number of C(H)(H)-C(H)(H)(H) pairs = 2; Number of
C(=O)-C(H)(H) pairs = 1; Number of C(=O)-N(H) pairs = 1; Number of C(H)(H)-N(H)
pairs = 1
If you left the box unchecked it would generate:
Number of C(H)(H)(CH3)-N(H) pairs = 1; Number of
C(=O)-N(H) pairs = 1; Number of C(=O)-C(H)(H)(CH3) pairs = 1
In a database containing all kinds of molecules
containing over 4000 values for log Kow, checking this box generated 400-500
unique combinations of pairs, while leaving the box uncheck generated over 1300
sub-structural features. Leaving the box
unchecked gives a model containing more chemical information, with the danger
that the model will be less general.
7. Minimum probability allowed. The default is set at
0.05, the typical value used by statisticians. This appears to work even with
data acquired from multiple sources from the internet. Setting this value
correctly is a function of the accuracy of your data, and setting it high or
low will affect where the analysis stops (how many independent variables are
included in the model.) For highly accurate data this value should be set very
low (molecular weight, for instance, could be set at p <=0.0001). For
sharing in the literature, you may find resistance from reviewers or regulators
with p > 0.05. [p = probability that an independent variable's correlation
is due to chance. 0.05 means 5% probability.]
8. Use connectivity and valence indices in the
analysis. This will cause the method to consider connectivity and valence
indices 1 through 4 and the kappa shape index 2 in the analysis. If will not,
though, force them into the model as an included variable. This feature is
useful if you think the model should contain both whole molecule properties and
sub-structural fragments in the model.
9. Include Hydrogen Bonding Interaction terms. The
program will consider some additional variables if you check this box. It will
look to see if each molecule contains more than one of the following features
and will create a cross product of the number of each pair in every molecule.
The H bonding groups it looks for are NH, NH2, OH, =O, COOH, SH, S(=O) and
P(=O). In our example of CH3CH2C(=O)NHCH2CH3 this would create one additional
term: NH x C=O. It would also create this term for the molecule O=CHCH2CH2NHCH3
(not just for amide.)
10. "Do not include molecules with formally
charged atoms." Checking this box will eliminate molecules with formally
charged atoms, for instance, Na+, Cl- from the analysis. It will eliminate most
salts, for instance, from the analysis of the chemelectrica.mdb and mydata.mdb
data sets.
11. Alternate
between forward and backward regression.
When this is checked the program will for do forward regression for a
number of steps. Next it will add all
the substructural features that had p<0.05 (or the significance selected in
step 7 above) to the model and do a backwards regression. It will continue until no variable with
p<0.05 or which increases r square by more than 0.0001 can be found and then
do a final backwards regression. The
program will do about 10 forward steps the first time, but thereafter will do
the number of forward steps you specify in the line after the check box
(default = 3 steps.) For large data
sets, we recommend you keep this box checked, because it greatly speeds the
analysis (answer in hours or days instead of weeks.)
If you do
not check this box, then the model will do a forward regression all the way to
the end and do a backwards regression only after no more significant variables
can be added. Leaving this box unchecked
can lead to a very long analysis with large, complex data sets (lasting weeks.)
12. Transform by dividing the variables by each
other. Checking this box will cause the
program to consider dividing every significant independent variable by each
other. In conjunction with repeating the
process (item 13 below) very complicated non-linear models are possible. It adds one to the denominator to avoid
division by zero. A consequence of this
is that it will also evaluate the
x/(x+1) transform (dividing a variable by itself plus one which
describes a curve converging on one.) Checking this option will greatly
increase the time of analysis and may make the model less general (while better
predicting that data in the database.)
With repeated iterations (next paragraph) it can be used to model very
complicated internal molecular interactions.
This feature is the one most likely to generate spurious correlations
(due to the number of possible variables being much greater than the number of
observations.)
13. Number of times to repeat the process (max 5): If
you check cross products and division, you will usually be able to get a better
model by repeating the process by going back to the step of doing a combination
of forward and backward regression, then repeating the combinations and
divisions steps. The process looks like
this:
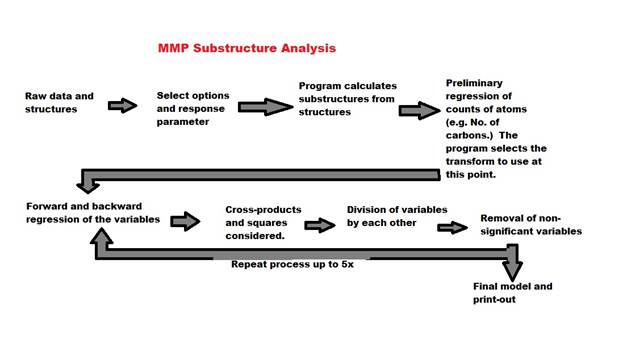
Usually two iterations are sufficient (keep the
default 2), but sometime three will do it, and one is often sufficient. Iterating the process will greatly lengthen
the time of the analysis. Variables can
become very complicated as cross product terms and division terms can be
further multiplied and divided by each other.
14.
Comments. If you would like to
append a line of comments to the output file (author, references, explanation),
place it here. For texts longer than a
sentence or so, create a separate document.
After hitting the "Done" button, a new
window appears from which you will select one response parameter (usually
experimental data you wish to analyze.) Select a parameter and hit
"Done."
Next a window will appear asking if you wish to force some
variables from your database into the analysis. This allows you to combine the
molecular properties calculated by MMP with sub-structural features of your
dataset.
Next you will see all the molecules in the database
flash by as the program determines the sub-structural features of the
molecules. The program is not looking for specific features, but creates the
sub-structural variables from scratch, depending on what combinations of atoms
are contained in all the molecules in the database. After completing this, the log window appears
and tells you how many molecules and variables will be included in the
analysis. The program next looks for fields created that have the same exact
values as other fields and for fields which have all the same values and eliminate
these variables from the dataset. The program will display the names of the
eliminated fields in the log window. The program proceeds with forward and
backwards stepwise regression, adding parameters to the model until (a) no
variables can be added that do not meet your significance level and the model r
square does not improve by at least 0.0001 or (b) you hit the Cancel button.
Cancelling goes to the next step in the analysis, not necessarily the end. After completion the model will do a final backwards
Stepwise regression to remove any variables that did not meet the level of
significance that you specified, one step at a time starting with the least
significant variable. If you selected
cross products and squared terms, it will then look at all the combinations of
the variables already in the model.
The program will ask you to save the dataset created
by the program to a tab delimited text file. This will save the molecule names,
the response (dependent variable), the predicted values, the molecular formula,
the molecular weight, the connection table link and all the independent
variable values. You can open up this database later in MMP or in Excel or any
other program that opens tab delimited text files. Note this data will not be
particularly secure. The program will also separately save all the information
in the log window to a file called mmp.txt and open it with Notepad. If you
want to save this log file save the file in Notepad and give it a different
name.
After saving the data the program will display the
model itself, as well as charts of predicted versus response and predicted
versus residual values. Residuals are response minus expected values. Clicking
on the data points to the far left or right of the residuals chart will
identify the outliers (poorly predicted molecules.) Often these outliers are
inaccurate experimental results. The file containing the model itself is
automatically stored in the mmp\models subdirectory. This file is parsed by the
program and should not be changed, though you can change the name of the file.
If you share this file with other people and they put it in their mmp\models
directory, then they will be able to use the file to predict properties of
molecules using MMP.
To predict the value of the property for a molecule
not in the database or for one which does not have an experimental value do the
following:
1. Draw the molecule.
2. In the Drawing window go to the Calculate menu and
choose "User Defined Properties." A new window will appear with a
list of user defined properties. Click on the one you want and hit Run.
NOTE: MMP+ comes with models for boiling point,
specific gravity and log Kow already in the \models folder. These models are
used by the program to calculate these properties, so over-writing or deleting
these files will change the default method used by MMP for calculating these
properties. If you are dissatisfied with the way MMP calculates these
properties you can re-run the analysis by opening the chemelectrica\mydata.mdb
database, adding or removing molecules, editing the literature data fields, and
re-running the substructure analysis. Running the log Kow analysis took about a
week on an Intel I5 processor.
COMMUNITY SHARING PAGE:
New models will be published periodically at www.norgwyn.com/models/Models.html. You can
download them there free of charge or submit new models for everyone to use
that will appear on this web page. From
the web page, just download the model text file into your mmp\models
subdirectory and they will be usable from the Calculate/User Defined Properties
menu of MMP.
A brief explanation of what is in the file for the
model:
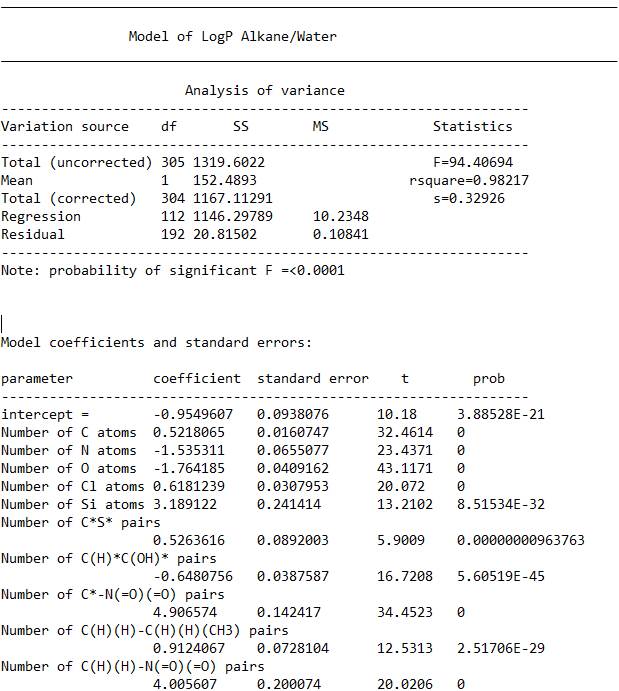
===============================================================
The Total (uncorrected) number of 305 is the number of
molecules contained in the final model. (df stands for degrees of freedom)
The number 112 next to “Regression” is the number of
variables. You usually want about 4x
more molecules than variables, depending on the quality of the data.
The larger the F score, the better the model. The probability of the F statistic being
significant is shown below the table.
R squared multiplied by one hundred is the percent of
the variance explained by the model.
S is the model standard deviation. The 95% confidence level is twice this, plus
or minus.
Below the analysis of variance table, you can see the
first terms in the model itself. The
format is, name of the variable, the coefficient that is multiplied by the
number of time the substructure appears in a molecule and the standard error, t
score and probability that the term is significant (0 is a term so significant,
the program could not determine the value.)
To predict the value of the property for a molecule
not in the database or for one which does not have an experimental value do the
following:
1. Draw the molecule.
2. In the Drawing window go to the Calculate menu and
choose "User Defined Properties." A new window will appear with a
list of user defined properties. Click on the one you want and hit Run.
NOTE: MMP+ comes with models for boiling point,
specific gravity and log Kow already in the \models folder. These models are
used by the program to calculate these properties, so over-writing or deleting
these files will change the default method used by MMP for calculating these
properties. If you are dissatisfied with the way MMP calculates these
properties you can re-run the analysis by opening the chemelectrica\mydata.mdb
database, adding or removing molecules, editing the literature data fields, and
re-running the substructure analysis. Running the log Kow analysis took about a
week on an Intel I5 processor.
2. Stepwise
Regression.
What this analysis will do: After you choose the
property to model, this method will attempt to correlate it with a combination
of all the other properties in your database. It will then use Stepwise
multiple regression to find the properties that are significantly correlated
with the response you choose and places the model in your \stepwise
subdirectory. You can then predict the property of any molecule you draw from
the Drawing window by selecting "user defined properties" from the
Calculate window.
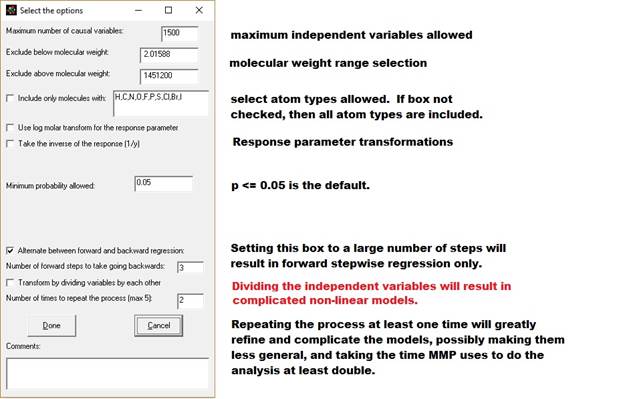
1. Maximum
number of causal variables: The program will calculate this based on the size
and complexity of your database, up the maximum allowed (see previous
paragraph). You can adjust this number here and in the Options menu.
2. Exclude by
molecular weight (boxes 2 and 3): You can limit the analysis to only consider
molecules between a minimum and maximum molecular weight.
3. Include
only molecules with: - Check this box if you want to only want to analyze
molecules containing specific atom types. You can modify which atoms you look
for by adding or subtracting atomic symbols from the comma separated list.
4. Use log
molar transform: After checking this box, your dependent variable (response
variable) will be transformed before analysis, i.e., y will become
log10(y/molecular weight). Note that the
data will be transformed back to the original transform when determining
properties of new molecules with unknown property values.
5. Take the
inverse of the response. After checking this box your dependent variable will
be transformed before analysis (i.e. y will become 1/y).
6. Minimum
probability allowed. I have set the default at 0.05, the typical value used by
statisticians. This appears to work even with data acquired from multiple
sources from the internet. Setting this value correctly is a function of the
accuracy of your data, and setting it high or low will effect where the
analysis stops (how many independent variables are included in the model.) For
highly accurate data this value should be set very low (molecular weight, for
instance, could be set at p <=0.0001). For sharing in the literature, you
may find resistance from reviewers or regulators with p > 0.05. [p =
probability that an independent variable's correlation is due to chance. 0.05
means 5% probability.]
7. Alternate between forward and backward
regression. When this is checked the
program will for do forward regression for a number of steps. Next it will add all the substructural
features that had p<0.05 (or the significance selected in step 7 aobove) to
the model and do a backwards regression.
It will continue until no variable with p<0.05 or which increases r
square by more than 0.0001 can be found and then do a final backwards
regression. The program will do about 10
forward steps the first time, but thereafter will do the number of forward
steps you specify in the line after the check box (default = 3 steps.) For large data sets, we recommend you keep
this box checked, because it greatly speeds the analysis (answer in hours or
days instead of weeks.)
If you do not check this box, then the
model will do a forward regression all the way to the end and do a backwards
regression only after no more significant variables can be added. Leaving this box unchecked can lead to a very
long analysis with large, complex data sets (lasting weeks.)
8. Transform
by dividing the variables by each other.
Checking this box will cause the program to consider dividing every
significant independent variable by each other.
In conjunction with repeating the process (item 13 below) very
complicated non-linear models are possible.
It adds one to the denominator to avoid division by zero. A consequence of this is that it will also
evaluate the x/(x+1) transform (dividing
a variable by itself plus one which describes a curve converging on one.)
Checking this option will greatly increase the time of analysis and may make
the model less general (while better predicting that data in the
database.) With repeated iterations
(next paragraph) it can be used to model very complicated internal molecular
interactions.
9. Number of
times to repeat the process (max 5): If you check cross products and division,
you will usually be able to get a better model by repeating the process by
going back to the step of doing a combination of forward and backward
regression, then repeating the combinations and divisions steps.
Usually two
iterations are sufficient (keep the default 2), but sometime three will do
it. Iterating the process will greatly
lengthen the time of the analysis.
Variables can become very complicated as cross product terms and
division terms can be further multiplied and divided by each other.
10. Comments.
If you would like to append a line of comments to the output file
(author, references, explanation), place it here. For texts longer than a sentence or so,
create a separate document.
After hitting
the "Done" button, a new window appears from which you will select
one response parameter (usually experimental data you wish to analyze.) Select
a parameter and hit "Done."
Next a window
will appear asking if you wish to force some variables from your database into
the analysis. This allows you to combine the molecular properties calculated by
MMP with sub-structural features of your dataset.
The list of
properties will appear again and this time you select properties to exclude
from the analysis (otherwise the program will look at every numeric field in
the database.)
The program
looks for fields created that have the same exact values as other fields and
for fields which have all the same values and eliminate these variables from
the dataset. The program will display the names of the eliminated fields in the
log window. The program proceeds with forward and backwards stepwise
regression, adding parameters to the model until (a) no variables can be added
that do not meet your significance level and the model r square does not
improve by at least 0.0001 or (b) you hit the Cancel button. After completion
the model will do a final backwards Stepwise regression to remove any variables
that did not meet the level of significance that you specified, one step at a
time starting with the least significant variable.
The program
will ask you to save the dataset created by the program to a tab delimited text
file. This will save the molecule names, the response (dependent variable), the
predicted values, the molecular formula, the molecular weight, the connection
table link and all the independent variable values. You can open up this
database later in MMP or in Excel or any other program that opens tab delimited
text files. Note this data will not be particularly secure. The program will
also separately save all the information in the log window to a file called
mmp.txt and open it with Notepad. If you want to save this log file save the
file in Notepad and give it a different name.
After saving
the data the program will display the model itself, as well as charts of
predicted versus response and predicted versus residual values. Residuals are
response minus expected values. Clicking on the data points to the far left or
right of the residuals chart will identify the outliers (poorly predicted
molecules.) Often these outliers are inaccurate experimental results. The file
containing the model itself is automatically stored in the mmp\stepwise
subdirectory. This file is parsed by the program and should not be changed,
though you can change the name of the file. If you share this file with other
people and they put it in their mmp\stepwise directory, then they will be able
to use the file to predict properties of molecules using MMP.
After the analysis is done, to predict the value of
the property for a molecule not in the database or for one which does not have
an experimental value do the following:
1. Draw the molecule.
2. In the Drawing window go to the Calculate menu and
choose "User Defined Properties." A new window will appear with a
list of user defined properties. Click on "Go to Stepwise Directory"
(unless already clicked.) Click on the property you want and hit Run.
NOTE: This
routine uses lots of RAM and lots of CPU time and can take a long time to
complete if you have a database of over 1000 molecules in it.
To access this feature: Open a database. Go to the
Analyze menu. Choose the second item, "Atom Based Analysis." This feature differs from the Substructure
Analysis in two ways. It (1) starts the
substructure from specific atoms using qualifications such a charge,
lipophlicity or location, while ignoring the other atoms AND (2) will create
substructures of many connected atoms, going more deeply into the
structures. For instance, you could
restrict an analysis of pKa to structure starting from the most positively charged
hydrogen atoms or you could restrict analysis of a polymer database to starting
with the two end atoms. You could model
a receptor by starting the substructures with up to 10 of the most negatively
charged atoms and include distances between the atoms from left to right along
the x axis.
Like the substructure analysis, this method will
create substructures from the molecules in your database which will be used as
independent variables in the analysis. This routine uses least squares multiple
regression to find the substructural features that are significantly correlated
with the property being modeled and places the model in your \atom_based
subdirectory. After completing the analysis, this property can be predicted for
any molecules you draw by selecting "user defined properties" from
the Calculate menu of the Drawing window.
What can be analyzed: This method does not care what
atom types you have in your database. It creates the substructures from the
molecules themselves, not from a look-up table. You should be able to analyze
any set of molecules as long as you have input the structures correctly.
Molecules should be drawn in a consistent manner (e.g. if you draw nitro as
N(=O)=O one time and N+(=O)O- the next it will not recognize them as the same
substructure. Salts and ionic molecules are also types of molecules that can be
drawn in many different ways.)
CAVEAT EMPTOR: On large databases this method
will take days to complete the task. On a database of 72 structurally related
molecules this method takes a few minutes to complete its task on a slow XP
computer. On a database of 800 molecules containing mostly solvents and
surfactants, this method took 2 hours to complete its task on a slow XP
computer. On a database containing over 4000 molecules with over 1300 unique
sub-structural features on an Intel I5, Windows 10 computer the program took
about a week to converge. Fortunately, the computer was still usable as the
task ran in the background. However, the database being used was tied up for
the duration of the analysis. The length of the task is a function of your
computing speed. The size of the database that can be used is limited by RAM.
If you expect to routinely use this program feature on large databases, we
recommend you have a computer dedicated to the task.
Details of the Method:
The number of variables that will be generated is a
function of the size and complexity of the structures in the database. The
default setting for maximum number of sub-structural variables is 1800. If you
get an "out of memory" error go to the Options menu of the Database
window and decrease the number of variables allowed in the analysis. If using
the mydata.mdb or chemelectrica.mdb database you will need to set this to about
2500 because the program will find over 2000 substructures with the atom depth
set to 4.
After selecting "Atom Based Analysis" from
the Analyze menu of the Database window the following Window will appear:
Details of the Method:
The number of variables that will be generated is a
function of the size and complexity of the structures in the database. The
default setting for maximum number of sub-structural variables is 1800. If you
get an "out of memory" error go to the Options menu of the Database
window and decrease the number of variables allowed in the analysis. If using
the mydata.mdb or chemelectrica.mdb database you will need to set this to about
2500 because the program will find over 2000 substructures with the atom depth
set to 4.
After selecting "Atom Based Analysis" from
the Analyze menu of the Database window the following Window will appear:
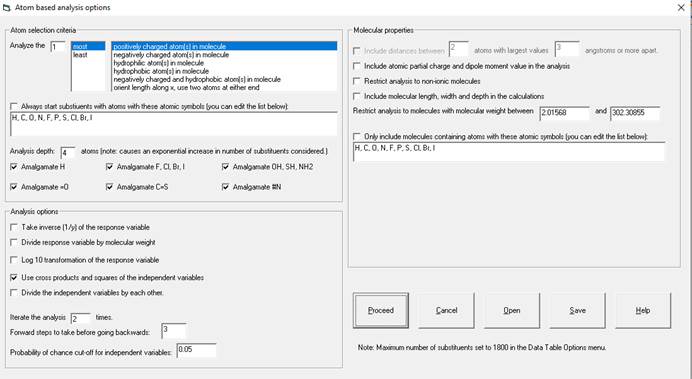
Explanation of this window follows:
Atom Selection Criteria:
1.
In the upper left
you choose what types of atoms to start the substructure generation and how
many. You can choose to analyze 1-10
atoms per molecule. You can choose
select starting atoms by charge, lipophilicity or the two atoms at the two ends
of the molecule oriented along the x axis.
Increasing the number of starting atoms will greatly increase the number
of variables (substructures) used in the calculations. As an example, if creating a model of lowest
pKa, if you have multiple atoms with pKa values (e.g., pKa 1, pka 2, etc.) you
may want to have 2 or more starting points so all are considered.
2.
You can restrict
the starting atoms by atomic symbols in a comma separated list. If you want to start the substructures with
hydrogen, for instance, you would check the box “Always start substituents with
atoms with these atomic symbols” and then change the text below this check box
to read “H”.
3.
Analysis depth
determines how deeply the program with delve into the molecule from the
starting point. For instance, if you
have opted to look at the one most positively charged hydrogen atom as your
starting point and have an analysis depth of 4, for n-hexanoic acid you would
generate the following substructures:
·
With Amalgamate
H, =O and OH checked: C(=O)(OH), C(=O)(OH)C(H)(H), C(=O)(OH)C(H)(H)C(H)(H), and
C(=O)(OH)C(H)(H)C(H)(H)C(H)(H).
·
With Amalgamate
H, =O and OH unchecked: H, OH, HOC, and HOC(=O)C(H)(H).
.
The depth you
select will greatly impact the time it takes to compute the model. Selecting a lesser depth will make your model
more generally applicable but decrease the accuracy for molecules in the
training set. In the literature, typical
models have a depth of 2 with amalgamation.
If your model has less than 4
molecules per variable, you may want to decrease the depth to make it more
generally applicable. If you only wish
to predict values for molecules with substructural features close to the
training set this may not be necessary and will decrease accuracy. If predictions made greatly vary from
experimental values, add the poorly predicted molecules to the database and
re-run the analysis.
.
Analysis Options:
1.
Check boxes for
transforming the dependent variable (the parameter being modeled.) can be
checked as desired. Your options are,
1/x, x/molecular weight and log10(x) or any combination of these three. Use one of these transformations if the
residuals have a pattern that looks like a triangle instead of pure randomness.
2.
Cross products
and squares of the independent variables (e.g. the substructures) will be
considered in the analysis (i.e. y1*y2,
y1*y1 etc.). Only the variables
found to be likely to be significant will be used.
3.
Divide the independent variables by each
other. Checking this box will cause the
program to consider dividing every likely-significant independent variable by
each other. In conjunction with
repeating the process very complicated non-linear models are possible. It adds one to the denominator to avoid
division by zero. A consequence of this
is that it will also evaluate the
x/(x+1) transform (dividing a variable by itself plus one which
describes a curve converging on one.) Checking this option will greatly
increase the time of analysis and may make the model less general (while better
predicting that data in the database.)
With repeated iterations (next paragraph) it can be used to model very
complicated internal molecular interactions.
This feature is the one most likely to generate spurious correlations
(due to the number of possible variables being much greater than the number of
observations.)
4.
Iterate the
process. If this number is greater than
one, the program will, after determining cross products and division go back to
the beginning of the analysis and repeat the process to see if there were any
significant variables missed before doing the cross-products, squares and
divisions.
5.
Forward step to
take before going backwards. The program
will do a standard forward stepwise regression for the selected number of steps
(more the first time through). Then it
will accelerate the analysis be adding all the significant independent
variables it found and do a backwards stepwise regression. Next it will do the forward regression and
repeat this process until there are no more variables with 3x of the selected
probability cut-off. At the end of the
analysis (including the cross products and divisions) all the variables still having a probability
above the cut-off (usually p=0.05) will be removed by a final backwards
regression.
6.
Probability of
chance cut-off for independent variables: The default is set at 0.05, the
typical value used by statisticians. This appears to work even with data
acquired from multiple sources from the internet. Setting this value correctly
is a function of the accuracy of your data, and setting it high or low will
affect where the analysis stops (how many independent variables are included in
the model.) For highly accurate data this value should be set very low
(molecular weight, for instance, could be set at p <=0.0001). For sharing in
the literature, you may find resistance from reviewers or regulators with p
> 0.05. [p = probability that an independent variable's correlation is due
to chance. 0.05 means 5% probability.]
Molecular Properties:
1.
Include distances
between [variable] atoms [variable) angstroms apart: This check box only is enabled when you
analyze more than one starting atom. It
will create independent variables for the distances between the starting atom
points.
2.
A) Include
partial charge and dipole moment in the analysis: This check box causes the partial charge of
the starting atoms to be included as an independent variable as well as the
molecular dipole moment as calculated by CNDO.
It is available if you used partial charge as the criteria for selecting
the starting atoms.
B) Include
atomic lipophilicity values in the analysis.
This check box causes the lipophilicity score of the starting atoms to
be included as an independent variable.
It is available if you use hydrophilicity or hydrophobicity as the
determinant for the starting atoms.
3.
Restrict
molecules to non-ionic molecules: Excludes all molecules with formal charges.
4.
Include molecular
length width and depth in the calculations:
Includes the length (along x axis), width (y axis) and depth (z axis) of
the molecules as independent variables in the analysis.
5.
Restrict
molecular weight to molecules between [lower bound] and [higher bound]: You can
limit the analysis to only consider molecules between a minimum and maximum
molecular weight. The default is the
full range of molecular weight present in the database.
Use Molecular Geometry in the Calculations:
These options will only be visible if more than one
atom is used as a starting point and the Include distances between atoms check
box under molecular properties is checked.
1. Sort atoms from left to right along the x axis. The starting atoms will be sorted by their
position along the x axis. If not
checked, the atoms will be sorted by how large the charge or hydrophobicity
score is. 2.
2. Orient molecules with maximum length along the x axis
and maximum width along the y axis: Before doing other calculations, each
molecular will be rotated so that the largest possible length and largest width
perpendicular to that length is made.
3. Rotate the molecule 180 degrees along Y is the X
component of dipole moment is negative and 180 degrees around the X axis if the
Y component of dipole moment is negative.
It does this AFTER rotating the molecular for maximum x and maximum y if
check box 2 above is checked. The check
box is only enabled if you select charge as the criteria for choosing the
starting atoms.
4. Rotate the molecule 180 degrees so that the most
lipophilic half is on the right side. It
does this AFTER rotating the molecular for maximum x and maximum y if check box
2 above is checked. The check box is
only enabled if you select charge as the criteria for choosing the starting
atoms.
5. Save changes in the atom’s orientation (i.e. if you
selected any of 2-4 above) to the database.
This may save time if you repeat the analysis. Note that if your molecular structures are
stored as separate files, the change in geometry will be saved to the files,
and made permanent. If, on the other
hand, you save the geometries within the database, then you must later save the
database to make the changes permanent.
After hitting the "Proceed" button, a new
window appears from which you will select one response parameter (usually
experimental data you wish to analyze.) Select a parameter and hit
"Done."
Next a window will appear asking if you wish to force
some variables from your database into the analysis. This allows you to combine
the molecular properties calculated by MMP with sub-structural features of your
dataset.
Next you will see all the molecules in the database
flash by as the program determines the sub-structural features of the
molecules. The program is not looking for specific features, but creates the
sub-structural variables from scratch, depending on what combinations of atoms
are contained in all the molecules in the database. After completing this, the log window appears
and tells you how many molecules and variables will be included in the
analysis. The program next looks for fields created that have the same exact
values as other fields and for fields which have all the same values and
eliminate these variables from the dataset. The program will display the names
of the eliminated fields in the log window. The program proceeds with forward
and backwards stepwise regression, adding parameters to the model until (a) no
variables can be added that do not meet your significance level and the model r
square does not improve by at least 0.0001 or (b) you hit the Cancel button.
Cancelling goes to the next step in the analysis, not necessarily the end. After completion the model will do a final
backwards Stepwise regression to remove any variables that did not meet the
level of significance that you specified, one step at a time starting with the
least significant variable. If you
selected cross products and squared terms, it will then look at all the combinations
of the variables already in the model.
The program will ask you to save the dataset created
by the program to a tab delimited text file. This will save the molecule names,
the response (dependent variable), the predicted values, the molecular formula,
the molecular weight, the connection table link and all the independent
variable values. You can open up this database later in MMP or in Excel or any
other program that opens tab delimited text files. Note this data will not be
particularly secure. The program will also separately save all the information
in the log window to a file called mmp.txt and open it with Notepad. If you
want to save this log file save the file in Notepad and give it a different
name.
After saving the data the program will display the
model itself, as well as charts of predicted versus response and predicted
versus residual values. Residuals are response minus expected values. Clicking
on the data points to the far left or right of the residuals chart will
identify the outliers (poorly predicted molecules.) Often these outliers are
inaccurate experimental results. The file containing the model itself is
automatically stored in the mmp\models subdirectory. This file is parsed by the
program and should not be changed, though you can change the name of the file.
If you share this file with other people and they put it in their mmp\models
directory, then they will be able to use the file to predict properties of
molecules using MMP.
To predict the value of the property for a molecule not
in the database or for one which does not have an experimental value do the
following:
1. Draw the molecule.
2. In the Drawing window go to the Calculate menu and
choose "User Defined Properties." A new window will appear with a
list of user defined properties. Click on the Atom Based button. Click on the property you want and hit Run.
COMMUNITY SHARING PAGE:
New models will be published periodically at www.norgwyn.com/models/Models.html. You can
download them there free of charge or submit new models for everyone to use
that will appear on this web page. From
the web page, just download the model text file into your mmp\models
subdirectory and they will be usable from the Calculate/User Defined Properties
menu of MMP.
MMP+ has integrated into it statistical tools used in
molecular design and QSPR (quantitative structure property relationships - the
study of the relationship of experimental properties of molecules to chemical
structure). QSAR (quantitative structure activity relationships) is the
application of these techniques to biological and pharmaceutical properties of
molecules, such as the inhibition of an enzyme important in some human
pathology. MMP+ also contains some
graphing and charting features used in this sort of research (see next section). When applied
to chemical data instead of biological response data QSAR is called QSPR
(quantitative structure-property relationships).
The statistical routines are accessed from the
Analysis menu of the Database Window. To
use the properties you must have first created a
database and opened
it. Alternatively you can use the
industrial.mdb database that comes with MMP+ for demonstration purposes. To learn about the data analysis features in
MMP+ open this database now (to open a database go to the File menu in the
Drawing Window, select Open Database, then choose ACCESS database from the file
types and open industrial.mdb in the MMP directory). The database window will
open as in Figure 12.
|
|
Figure 12.
Database window showing the Analyze menu. The industrial.mdb ACCESS database that
comes with the program has been opened. |
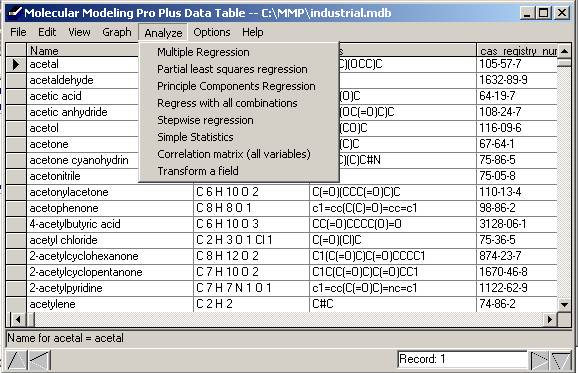
In this initial analysis we are going to use standard
multiple regression analysis. Multiple
regression is sensitive to the number of compounds relative to the number of
independent variables. Most QSAR/QSPR
data sets are characterized by containing approximately 15 compounds. In order to eliminate the possibility of
chance correlations, no more than 3 independent variables should be considered
for regression in the initial phase of the study. When these conditions exist, multiple
regression is the method of choice for the QSAR study. Otherwise you may want to use partial least
squares regression (part 3).
For purposes of demonstration we are going to model
the field Flash Point, which contains approximately 360 experimentally
determined values.
There are several possible ways to start the modeling
process. If you have no clue where to
start, the Regress with all combinations, the Stepwise regression procedure and
the partial least square (PLS) routines can all be considered. In this demonstration we will use linear
least squares regression and the routine Regress
with all combinations. Select this
option from the Analyze menu and check the two boxes so the options panel
appears as in Figure 13.
|
|
Figure 13.
Options panel for the Regress
with all combinations routine. We
will look at all 1, 2 and 3 variable combinations of variables and look at
transforming both the result parameter (Flash Point) and the causal
parameters. |
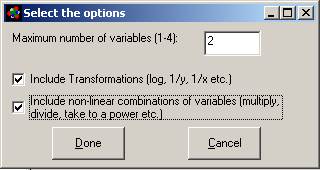
A list of variables appears and the program asks for
the Response parameter. Choose
Flash_point_C from near the bottom of the list (and hit the Done button). The list of fields appears again, this time
asking for parameters to include (force into the model). Just hit Done without selecting anything (no
included variables). The list of fields
appears a third time, but this time asks if you want to exclude anything
(figure 14). Click on the fields starting with name and ending with Trade
Names. Also select the fields starting
with Solvochromatic_cavity_term and ending with the last field,
connection_table_name as we do not want to include the experimental,
non-calculated fields.
|
|
Figure 14.
The field list in the Regress
with all combinations routine. The
example shows exclusion of text fields
not to be part of the analysis. |
The procedure will begin its calculations, considering
every combination of parameters. Since
we checked off the transformation and cross-products boxes its will also
consider transform of both the result parameter (dependent variable) and the
causal parameters (independent variables).
The top 50 one-variable models, transformed variable model,
cross-product models and two variable models (including transforms) will print
out. Below is an abbreviated version of
this print-out with only the best 5 models in each category listed (instead of
50).
|
Molecular Modeling Pro Plus 2004, Norgwyn
Montgomery Software Inc. One variable models for Flash_Point_(C) Variable df Intercept Coefficient r squared probability Critical_pressure_(bar) 360 215.768
-3.97404 0.56163 << 0.00001 ----- surface_tension_in_water 360 -541.294 23.9295 0.546262 << 0.00001 ----- boiling_point 360
47.3372 0.147412 0.461673 << 0.00001 ----- water_solubility 360
93.4639 -12.3763 0.388525 3.03687E-40 ----- Log_P 360 66.8162 8.48604 0.289003 1.99958E-28 ----- ==================================================
Transformed variable models for
Flash_Point_(C) Variable df Intercept Coefficient r squared probability x transform:Flash_Point_(C) 1/Enthalpy_of_vaporization_at_the_boiling_point_(kJ/mole)
360 261.011
-7787.1 0.744842 << 0.00001 ----- x transform:Flash_Point_(C) 1/ln(connectivity_1) [+4.86681610107422] 360 459.642
-860.804 0.7332 << 0.00001 ----- x transform:Flash_Point_(C)^2 1/ln(kappa_2) [+8.8418798828125] 360 123880
-297587 0.718872 << 0.00001 ----- x transform:Flash_Point_(C)^2 1/ln(connectivity_1) [+4.86681610107422] 360 116635
-232747 0.716677 << 0.00001 ----- x transform:Flash_Point_(C)^2 1/ln(log_olive_oil_gas_partition_coefficient)
[+25.8770092773437] 360 167359
-613474 0.714944 << 0.00001 ==================================================
Cross products, divisions and subtractions
of 2 variables for Flash_Point_(C) Variable df Intercept Coefficient r squared probability 1/Enthalpy_of_vaporization_at_the_boiling_point_(kJ/mole)*1/ln(connectivity_1)
[+4.86681610107422] 360 236.507
-14276.4 0.743771 << 0.00001 ----- 1/Enthalpy_of_vaporization_at_the_boiling_point_(kJ/mole)*ln(boiling_point)
[+55.0082995986938] 360 284.957
-1693.46 0.740526 << 0.00001 ----- surface_tension_in_water*ln(boiling_point)
[+55.0082995986938] 360 -252.301 2.2992 0.734733 << 0.00001 ----- surface_tension_in_water*1/Enthalpy_of_vaporization_at_the_boiling_point_(kJ/mole)
360 272.602
-324.658 0.723927 << 0.00001 ----- LUMO-surface_tension_in_water 337 -696.529
-29.7928 0.716809 << 0.00001 ----- ==================================================
Two variable models for Flash_Point_(C) Variable df Intercept Coefficient r squared probability 1/Enthalpy_of_vaporization_at_the_boiling_point_(kJ/mole)*1/ln(connectivity_1)
[+4.86681610107422] 360 271.411
-24629.7 0.808152 << 0.00001 1/Enthalpy_of_vaporization_at_the_boiling_point_(kJ/mole)*1/ln(connectivity_1)
[+4.86681610107422]^2
510166 ----- 1/Enthalpy_of_vaporization_at_the_boiling_point_(kJ/mole)
337 -231.512
-4543.29 0.802904 << 0.00001 LUMO-surface_tension_in_water -15.9342 ----- surface_tension_in_water*1/Enthalpy_of_vaporization_at_the_boiling_point_(kJ/mole)
337 -248.001
-187.422 0.801363 << 0.00001 LUMO-surface_tension_in_water -16.7771 ----- 1/Enthalpy_of_vaporization_at_the_boiling_point_(kJ/mole)
337 -230.23
-4639.37 0.800536 << 0.00001 HOMO-surface_tension_in_water -15.5848 ----- 1/Enthalpy_of_vaporization_at_the_boiling_point_(kJ/mole)*ln(boiling_point)
[+55.0082995986938] 337 -207.114
-1023.02 0.80015 << 0.00001 LUMO-surface_tension_in_water -15.7089 ==================================================
Two variable models for Flash_Point_(C)^2 Variable df Intercept Coefficient r squared probability 1/Enthalpy_of_vaporization_at_the_boiling_point_(kJ/mole)*1/ln(connectivity_1)
[+4.86681610107422] 360 67751.9
-7887220 0.756959 << 0.00001 1/Enthalpy_of_vaporization_at_the_boiling_point_(kJ/mole)*1/ln(connectivity_1)
[+4.86681610107422]^2
215334000 ----- surface_tension_in_water 360 29844.4 2420.96 0.755017 << 0.00001 1/ln(connectivity_1) [+4.86681610107422]
-180281 ----- surface_tension_in_water 360 28059.2 2571.22 0.751007 << 0.00001 1/Enthalpy_of_vaporization_at_the_boiling_point_(kJ/mole)-1/ln(connectivity_1)
[+4.86681610107422]
195313 ----- boiling_point 337
-144783 29.222 0.749497 << 0.00001 LUMO-surface_tension_in_water -5805.75 ----- boiling_point 337
-146922 30.2855 0.74891 << 0.00001 HOMO-surface_tension_in_water -5735.66 ----- |
|
Figure 14.
Print-out from the Regress all combinations routine (options selected:
2 independent variables, with transformations and cross-products). See discussion. |
The Regress all
combinations routine gives us needs to be used with a degree of common
sense. It is only the first step in the
analysis of the data. In this real world
example, none of the one variable models is particularly satisfying. The best one variable model is:
Flash point (C)
= 215.768 + -3.97404*Critical_pressure_(bar)
N=360, r squared = 0.56163, p << 0.00001
Although from the probability (<<0.00001) we can
deduce that their is a significant relationship between flash point and
criticial pressure, it does not make good chemical sense that critical pressure
is the main underlying cause in the variance in flash point. It is more likely that they have, in part,
the same underlying cause, and are therefore inter-correlated, but not cause
and effect. The same can be said of all
the other one variable models. The other
statistics, N is the degrees of freedom (number of data points - number of
independent variables), and r squared is the proportion of the variance
accounted for by the model (i.e. in this example approximately 56%, while 44%
is still unaccounted for and in the model error term). The p <<0.00001 term indicates that
there is less than an 0.001% chance that the relationship is strictly due to
chance.
The best transformed model is much more satisfactory
from a chemical stand-point.
Flash point (C)
= 261.011 + -7787.1*1/Enthalpy_of_vaporization_at_the_boiling_point_(kJ/mole)
N = 360, r
squared = 0.744842, p << 0.00001
This model also accounts for more of the variance in
Flash point (74.5%).
The next section has models that are combinations of
two variables (multiply them, divide them, subtract them). These models are more complicated then the
transformed enthalpy of vaporization model and do not explain any more of the
variance. Because of this, we will
tentatively choose the 1/enthalpy of vaporization model as better than these
models. The best of these models is:
Flash point (C) = 236.507 -
14276.4*(1/Enthalpy_of_vaporization_at_the_boiling_point_(kJ/mole)*1/ln(connectivity_index_1
+ 4.87) )
N = 360, r squared =
0.743771, p << 0.00001
The best two variable model is a parabolic version of
the cross-product model above:
Flash point (C) = 271.411
-
24629.7*(1/Enthalpy_of_vaporization_at_the_boiling_point_(kJ/mole)*1/ln(connectivity_1+4.87))
+
510166*(1/Enthalpy_of_vaporization_at_the_boiling_point_(kJ/mole)*1/ln(connectivity_1+4.87))^2
N = 360 , r squared = 0.808152, p << 0.00001
Since this model is a fairly significant improvement,
it will come under consideration as we develop the model. It will be compared with the simpler one
variable transformed model (1/enthalpy of vaporization) as we refine the
model. Connectivity index 1 is a measure
of molecular size. So this model implies
a parabolic relationship between the inverse of the enthalpy of vaporization
multiplied by the inverse of the log of molecular size. In other words the lower the enthalpy of
vaporization and the lower the molecular size, the higher the flash point.
The final section of results are for models of the
square of the flash point, which was the best dependent variable transform
found. However none of these models
explain as much of the variance as the two variable model for the untransformed
values of flash point, so we can discard them.
At this point, we will graph the data (next section)
and further refine the models (Analyze Data, part 2).
Graph Data
Again, if you want to follow along and do what is
described open the industrial.mdb database that comes with the program. Go to
the database window. Go to the Graph
menu and select XY(2-D) plots/Simple. For the y (response) axis select
Flash_Point_C from the list that appears and for the x axis select
enthalpy_of_vaporization_at_the_boiling_point_kj/mole.
|
|
Figure 15. The "Simple" x-y plot. Its one
advantage over the advanced plot is that you can click on a data point with
the mouse and the data point is identified (in this example the arrow points
to PPG-24-buteth-27). This ability
make it easy to identify outliers not described by the model and wrongly
entered data points. |
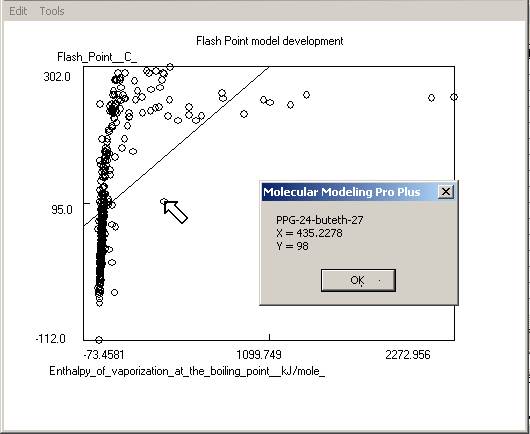
You can clearly see in figure 15 that the flash point data
is poorly modeled by a straight line regression to enthalpy of
vaporization. However, we know from the
analysis in the previous section that there is a much better relationship
between flash point and the inverse of enthalpy of vaporization. We can see this relationship using the
advanced graphing feature.
Go to the Graph menu.
Select XY (2-D) plots/Advanced.
For the y axis select Flash_Point_C from the list that appears and for
the x axis select enthalpy_of_vaporization_at_the_boiling_point_kj/mole. Answer the question about wanting a second y
axis "No". The options panel
appears as in figure 16. Fill in the
options as shown.
|
|
Figure 16.
The options panel for the "advanced" 2-D XY plot. We are going to color the plot by a third
variable (connectivity index 1). The
minimum and maximum x and y values have been rounded for better looking graph
labels. We also are requesting that
the x axis undergo the 1/x transform. |
Hit the done button in the options panel. The list of fields again appears so you can
select the field to color the data points by.
Select connectivity_1 from the list.
The plot in figure 17 appears.
|
|
|
|
Figure 17.
Example of the XY (2-D) "advanced" plot. Flash point is fairly well correlated with
1/enthalpy of vaporization (the curve drawn through the data points is that
made by the linear least squares regression model). Data points are colored by a third variable
(connectivity index 1). It appears
that enthalpy of vaporization and connectivity index 1 are probably also
intercorrelated as the colors of the data points are clustered. The model flash point = 1/enthalpy of
vaporization accounts for about 75% of the variance for 360 values of flash
point of solvents and surfactants found in the industrial.mdb database. Most of the values with lower connectivity
indices (dark blue) are solvents and are found toward the left side of the
graph. Their behavior is different
from surfactants. The Joback and Reid
enthalpy of vaporization calculation was built with solvents well-represented
and surfactants not represented. A
question to ask about this model is that is the inverse transform really
needed or is it reflecting inaccuracy in the calculation of enthalpy of
vaporization. Note that a very
straightforward linear relationship appears to exist between flash point and
enthalpy of vaporization for solvents.
These sorts of insights can only be found by plotting the data. |
|
From figure 17 we note that The PPG-24-buteth-27 point
previously noted in figure 15 is an outlier.
There is quite a bit of "slop" near the inflection point of
the curve as well. Most of the data
points with a low enthalpy of vaporization are tightly clustered in a linear
relationship with flash point. A question
that should be asked is if there should not be two separate models. One for small molecules with low connectivity
indices (dark blue data points) and a second model for the rest.
Next let's investigate the two variable models using
the standard 3-D chart. With the
solubility database open and in the Database Window go to the Graph Menu and
from the 3-D plot menu choose 3-D Graph.
When the program asks for the y (response) parameter choose flash point. For the first x (causal parameter) choose enthalpy_of_vaporization_at_the_boiling_point_(kg/mol)
and for the second x parameter select connectivity_1. Next comes the options window. Under y (response parameter) leave all the
transformation boxes unchecked. Under
x1, check 1/x1. Also check the "Use
the square of the causals" box.
|
|
Figure 18.
The 3-dimensional plot options box. |
Running this routines with the settings in figure 18
generates the analysis of variance chart in figure 19 and the 3-D plot in
figure 20 as well as a plot of the model predicted vs. actural values and model
predicted vs. residuals (not shown).
|
Analysis of variance ------------------------------------------------------------------
Variation source df
SS MS Statistics ------------------------------------------------------------------
Total (uncorrected) 361
6978060.7389 F=304.47018
Mean 1 3592866.3781 rsquare=0.8109 Total (corrected) 360 3385194.3608 s=42.46382 Regression 5
2745066.8301 549013.36602 Residual 355 640127.53069 1803.17614 ------------------------------------------------------------------
Note: probability of
significant F =<0.0001 Model coefficients and
standard errors: parameter coefficient standard error t
prob ------------------------------------------------------------------
intercept = 340.345 12.8173 26.5534 0 1/Enthalpy_of_vaporization_at_the_boiling_point_(kJ/mole)
-15998.7 999.898 16.0004 6.5861E-44 connectivity_1 -0.7123 0.17697 4.02498 0.0000694934 1/Enthalpy_of_vaporization_at_the_boiling_point_(kJ/mole)*connectivity_1
42.1873 5.69143 7.41244 8.93892E-13 1/Enthalpy_of_vaporization_at_the_boiling_point_(kJ/mole)^2
175578 19755.1 8.88776 3.0396E-17 connectivity_1^2 0.00115334 0.000391426 2.94649
0.00342328 ------------------------------------------------------------------
note: response variable:
Flash_Point_(C) |
|
Figure 19.
Example of the Analysis of variance table and model printed out by the
3-dimensional plotting routines. |
In this analysis we are comparing a parabolic model
with the two variable model found in part one.
Here are the two models side by side:
Two variable model from part 1.
Flash point (C) = 271.411
-
24629.7*(1/Enthalpy_of_vaporization_at_the_boiling_point_(kJ/mole)*1/ln(connectivity_1+4.87))
+
510166*(1/Enthalpy_of_vaporization_at_the_boiling_point_(kJ/mole)*1/ln(connectivity_1+4.87))^2
N = 360 , r squared = 0.808152, p << 0.00001
Parabolic model (figure 19):
Flash point (C) = 340.345 +
-15998.7*(1/enthalpy of vaporization) +
-0.7123*connectivity index 1
+42.1873*((1/enthalpy of vaporization) * connectivity
index 1) +
+175578*(square of 1/enthalpy of vaporization) +
+0.001155334*(square of conntectivity index 1)
N=360, r
squared = 0.81, model and all variables highly significant
Which model is to be preferred? The first model contains fewer terms with
about the same r squared value and is probably the one to use. However, as we develop the model, it will be
good to keep in mind that we may be able to substitute in the parabolic model
if the log(connectivity index 1) cross product begins to look suspect. Below
(figure 20) is the response surface for the parabolic model.
|
|
|
|
Figure 20.
Response surface generated by the 3-D chart routine for the flash
point model discussed above. It is
evident from the plot that flash point rises with increasing connectivity
index 1 and decreasing 1/enthalpy of vaporization. The actual model used to construct this
surface is detailed in figure 19. |
|
An alternate way of displaying this same data is the
contour plot. To generate a contour plot
expressing this same flash point model of the data in the industrial.mdb go to
the Graph menu. Choose Contour plot from the 3-D Plot menu. When the program asks for
the y (response) parameter choose flash
point. For the first x (causal
parameter) choose enthalpy_of_vaporization_at_the_boiling_point_(kg/mol)
and for the second x parameter select connectivity_1. Next comes the options window. Under y (response parameter) leave all the
transformation boxes unchecked. Under
x1, check 1/x1. Also check the "Use
the square of the causals" box. The options panel will again look like
figure 18 except that we will accept the default graph title. This generates the same statistics (figure
19) and will generate the plot in figure 21 instead of figure 20. If you will compare figures 20 and 21 you
will note that they really contain exactly the same information.
|
|
|
Figure 21. The
flash point model depicted as a contour plot.
The same information is contained in figure 20. |
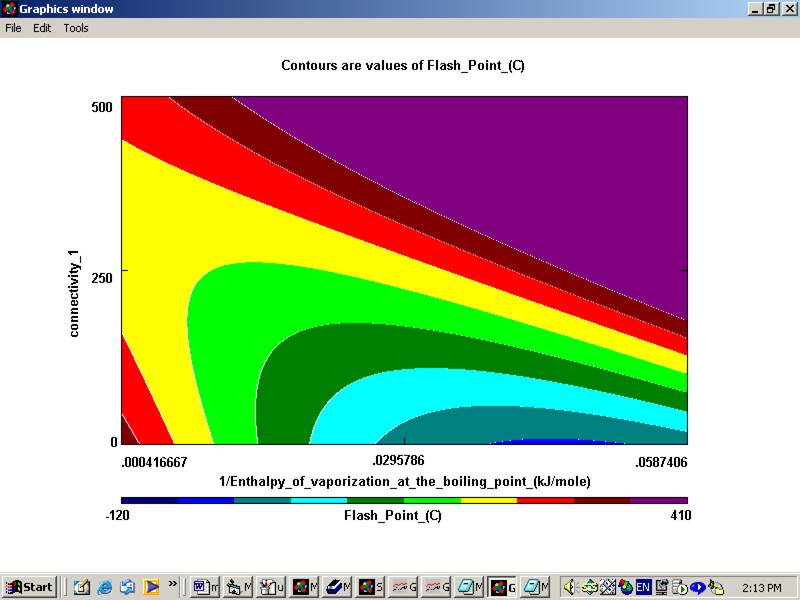
The last type of plot is a rotating 3-D plot. Go to the Graph menu. From the 3-D
plot menu item select Rotatable 3-D
Plot. From the lists that appear select the same 3 fields as in the other
examples (enthalpy of vaporization at the boiling point (kg/mole) flash point
(C) and connectivity 1). The plot in
figure 21 appears.
|
|
Figure 22.
Investigating the relationship between flash point, enthalpy of
vaporization and connectivity index 1 using the rotating 3-D plot. This plot can be rotated about and the data
points colored by values of a field to look for relationships in 3 and 4
dimensions. |
Use the rotate menu to rotate the plot. You can freeze the rotation at any point by
holding the mouse button down. Look for
linear relationships, clusters of points and other features that when explained
will give insight into the relationships between the variables.
The analysis of the flash point model continues in the
next two sections.
In this section we are going to critically examine the
models generated in part 1 with the brute force Regress all variables routine.
After doing this, we will briefly discuss what the next steps are beyond
the one and two variable models we found in part one (i.e. looking at more
complicated models). Some statistical
training will be helpful in this section.
Creation of transformed fields:
Open the industrial.mdb database that comes with the
program. Go to the database window. The first thing we are going to do is create some
new fields. Have patience here as some
of these steps will take the computer a few minutes to complete. The first new field we will create is
1/enthalpy of vaporizatiion.
Go to the database Window. Go to the Analyze menu. Select Transform
a Field. The options panel in figure 23 will appear. Select Enthalpy of vaporization... from the
list at the left and 1/(enthalpy of
vaporization... as shown.
|
|
Figure 23.
The field transform options panel. |
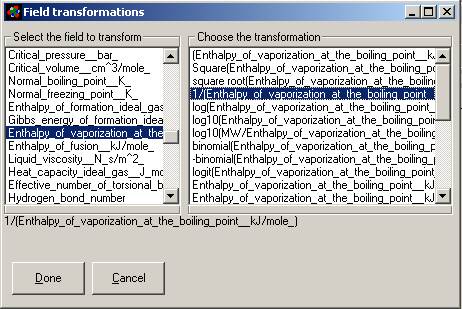
When the program asks which field to store the new
field end, scroll to the bottom of the list and select "New
Field." Wait. Save the database (File, Save) to the
Microsoft Access format using the file name industrial2.mdb.
Next we create the more complicated transformed field
used in the two variable model:
(1/enthalpy of vaporization)*(1/(ln(connectivity index
1 + 4.866816017422))
Creating this field is a multistep process using the
Analyze menu/Transform a field routine:
1. Create a field that adds 4.866816017422 to
connectivity_1
2. Create a field containing the log of the field
created in step 1
3. Create the inverse (1/x) of the field created in
step 2.
4. Multiply the field created in step 3 with
1/enthalpy of vaporization created in the first example (figure 23).
5. Save to industrial2.mdb.
Multiple Regression Analysis with
PRESS:
One variable model:
Let's look at the best one variable model from Part 1
in more detail (flash pt. = b0 + b1*1/enthalpy of vaporization). Go to the Analyze menu. Select Multiple
Regression Analysis. When the
program asks for the y (response) variable choose Flash Point (C) off the list
and check the box labeled Conduct PRESS
Analysis.
|
|
|
|
Figure 24.
Select y (dependent variable) from the list and check the PRESS
analysis box and hit Done. |
Figure 25.
Select x (independent variable) from the list and hit Done. |
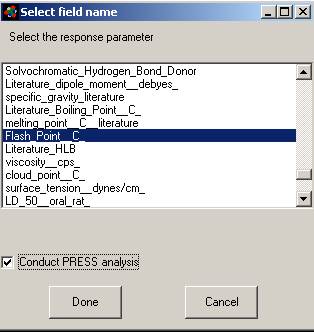
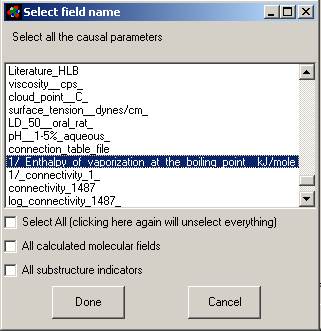
Next select 1/enthalpy of vaporization from the causal
parameter list. The following analysis
will appear. Explanations and analysis
appear in the figure legends.
|
Analysis of variance ------------------------------------------------------------------
Variation source df
SS MS Statistics ------------------------------------------------------------------
Total (uncorrected) 360 6818859.7389 F=1066.68276 Mean 1 3523457.24432 rsquare=0.74872 Total (corrected) 359 3295402.49458 s=48.09447 Regression 1
2467320.53578 2467320.53578 Residual 358 828081.9588 2313.0781 ------------------------------------------------------------------
Note: probability of significant F
=<0.0001 Model coefficients and standard errors: parameter coefficient standard error t
prob ------------------------------------------------------------------
intercept = 259.139 5.52153 46.9325 <<0.00001 1/_Enthalpy_of_vaporization_at_the_boiling_point__kJ/mole__
-7722.05 236.437 32.6601 <<0.00001 ------------------------------------------------------------------
note: response variable: Flash_Point__C_ Printout of response values, predicted
values and residuals: observed predicted residual acetal
-21 27.8085 -48.8085 acetaldehyde -40 -29.363 -10.637 acetic acid 40 51.613 -11.613 acetic anhydride 54 62.7094 -8.7094 acetol 56 90.1554 -34.1554 acetone -17 -6.97324 -10.0268 acetone cyanohydrin 63 105.799 -42.7992 ...(and so on)... |
|
Figure 26.
Analysis of variance table, model coefficients and partial print-out
of the table of response, predicted and residual values for the flash point
one variable model. Analysis of variance table: Abbreviations used: df = degrees of freedom; SS =
sum of squares; MS = mean squared; F= Fischer's F test; r squared =
proportion of variance accounted for by the model (e.g. in this example about
75%); S = model standard deviation (about 95% of the data lies within 2
standard deviations - thus plus or minus about 96 degrees C); Coefficients table: The model is
-- : flash point = 259.1 - 7722.05*(1/enthalpy of vaporization) Both the intercept and the regression coefficient
are highly statistically significant (prob <<0.00001) Printout of predicted and residual values: From this table find the largest out-liers
(residuals) and determine if they have something obviously in common that
will lead to a better model. For
instance, if all solvents are well-accounted for, but surfactants are poorly
predicted, consider developing separate models for solvents and surfactants. |
|
Contributions to PRESS (Predictive Residual
Sum of Squares):
Compound Predictive
discrepancy -------------------------- ---------------------- acetal 2405.442 acetaldehyde 115.3149 acetic acid 135.8609 acetic anhydride 76.35809 acetol 1173.177 acetone 102.0248 acetone cyanohydrin 1842.063 acetonitrile 1.354055 ...(and so on)... Total PRESS = 843833.3 Sum of squares of response (SSY) = 8277222 Press/SSY = 0.1019464 The model appears to be reasonable and has
passed the cross-validation test. |
|
Figure 27.
PRESS output for the one-variable flash point model. The larger the predictive descrepancy, the
more influential the data point.
Leaving out an influential data point can greatly effect the
model. You may want to leave out some
of the more influential point and rerun the analysis. Also, as with the table of residuals
(figure 26) you may want to look for obvious patterns in the types of
chemistries that are influential. At the bottom of the table is an assessment of
whether the model meets the Press/SSY <= 0.4 cut-off. Failure would mean that just a few data
points are contributing to most of what is accounted for by the model and you
may want to get rid of some of these data points and redo the whole analysis.
|
|
|
Figure 28.
Table of observed versus predicted values of flash point in the one
variable model. Outliers (data points
farthest removed from the regression line) can be identified by clicking on
them on the graph with the mouse.
Make sure that outlying data points are not typos (check for errors). Consider whether the calculated field
(1/enthalpy of vaporization) is likely to calculate the outlying data points
well and if not exclude them from the analysis and tell the users of the
model that it is not reliable for those types of molecules. |
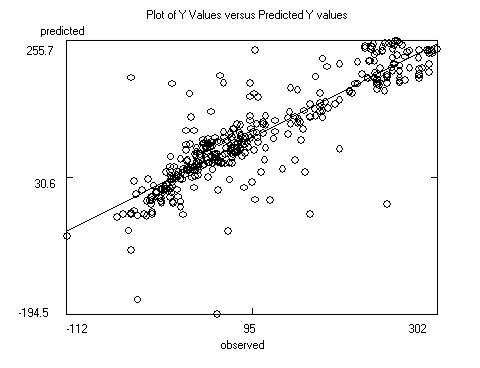
|
|
Figure 29.
Plot of predicted values of flash point against the residual values
(residuals = difference between predicted and observed values). Optimally this plat is a scatter plot with
completely random arrangement of the data points. Curvature or patterns may indicate problems
like intercorellation of the dependent and independent variables or the need
for transformation of one of the fields or addition of a parabolic or
cross-product term. Examination of the
residuals is a critical part of model validation. Compare with figure 32 and
note the further discussion there. |
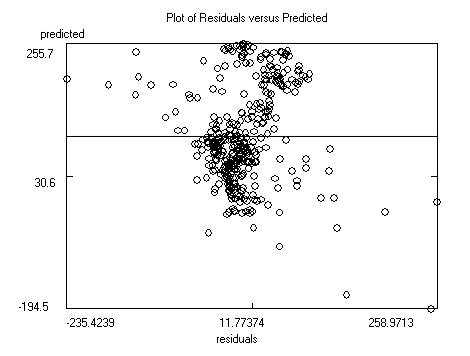
A usual follow-up here would be to look at the second
and third best one variable models (or some other model which makes good
intuitive chemical sense). Particularly
we would want to be aware if these other models have one or two very large
out-lying data points which if removed would give a model superior to the
1/enthalpy of vaporization model. If
they did it is likely that we would prefer one of these other models,
particularly if on examination the out-lying data points have a logical reason
for exclusion.
Two variable model:
|
Analysis of variance ------------------------------------------------------------------
Variation source df
SS MS Statistics ------------------------------------------------------------------
Total (uncorrected) 360 6818859.7389 F=770.74194 Mean 1 3523457.24432 rsquare=0.81196 Total (corrected) 359 3295402.49458 s=41.66302 Regression 2
2675719.231 1337859.6155 Residual 357 619683.26358 1735.80746 ------------------------------------------------------------------
Note: probability of significant F
=<0.0001 Model coefficients and standard errors: parameter coefficient standard error t
prob ------------------------------------------------------------------
intercept = 269.413 5.25699 51.2485 <<0.00001 1/_Enthalpy_of_vaporization_at_the_boiling_point__kJ/mole___plus
-24378.4 998.955 24.4039 <<0.00001 Square_1/_Enthalpy_of_vaporization_at_the_boiling_point__kJ/mole
503119 45561.2 11.0427 1.326E-24 ------------------------------------------------------------------
note: response variable: Flash_Point__C_ Total PRESS = 631855.3 Sum of squares of response (SSY) = 8277222 Press/SSY = 7.633664E-02 The model is excellent and passed the
cross-validation test easily. |
|
Figure 30.
Two variable model of Flash point.
See figure 26 for an explanation of abbreviations used in the analysis
of variance table. The two variable model explains about 6% more of the
variance in the table compared to the one variable model in figure 26 (e.g. r
squared = 0.81 vs. r squared = 0.76) and has a significantly lower model
standard deviation (S = 41.66). For a
data set of this size (df = 359) this is a significant and all things being
equal, this model is preferred over the one variable model. The PRESS/SSY cross-validation value has
improved substantially as well. In the
coefficients table check to see that all coefficients are statistically
significant (they are). |
|
|
Figure 31.
Plot of observed versus predicted values for the two variable model
for flash point. About 90% of the data
points lie quite close to the regression line. However the other 10% are poorly accounted
for. There are several possible
explanations. (1) wrong selection of
independent variables; (2) need more terms in the model; (3) poor accuracy in
flash point determination; (4) poor accuracy in enthalpy of vaporization
calculation; (5) some molecule types fit in the model and some don't (examine
the data points for this). |
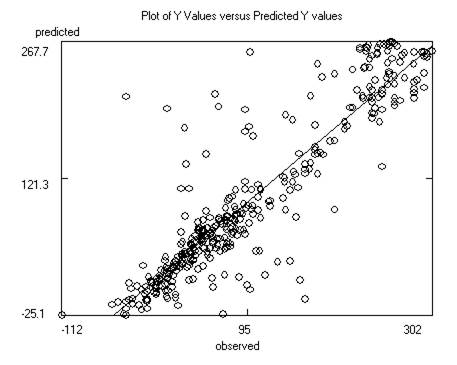
|
|
Figure 32. Plot
of predicted versus residual values for the two variable flash point model.
Data points on the far left or right of the graph are out-liers. The well
accounted for points are a scatter plot, but the out-liers form a pattern
going from the upper left to the lower right of the screen (i.e. as the
predicted value rises, the residuals become more negative). This indicates that there is likely another
term that needs to be added to the model or that the field transformation we
have selected is not the right one. |
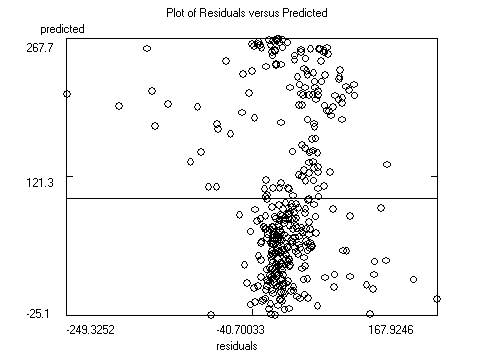
What next?
The analysis presented here is only the first
step. The next step is to build on what
we have found by going beyond 2 variable models. We have plenty of degrees of freedom left
(360) and with such a large data set
could comfortably add many variables.
A rule of thuimb is that there should be at least 8 molecules per
variable, although it depends on what purpose the data is being used whether
you can accept more or fewer variables.
The process is to include the variables found in the
one and two variable models above in all the more complicated models we
build. For instance, we can look for 2
and 3 variable models of flash point in which one of the variables is always
1/enthalpy of vaporization. The analysis
is done exactly as in Part 1 with the Regress
all combinations routine, except when the program asks for included
variables where previously we checked nothing, we now check 1/enthalpy of
vaporization (results, figure 33).
|
Molecular Modeling Pro Plus 2004, Norgwyn
Montgomery Software Inc. Variables included in every model are:
1/_Enthalpy_of_vaporization_at_the_boiling_point__kJ/mole__ One variable models for Flash_Point__C_ Variable df Intercept Coefficient r squared probability 1/_Enthalpy_of_vaporization_at_the_boiling_point__kJ/mole__
359 -0.0249783
-6095.31 0.784338 << 0.00001 surface_tension_in_water 8.41897 ----- 1/_Enthalpy_of_vaporization_at_the_boiling_point__kJ/mole__
359 221.921
-7059.8 0.778774 << 0.00001 percent_hydrophilic_surface 0.575727 ----- 1/_Enthalpy_of_vaporization_at_the_boiling_point__kJ/mole__
359 278.799
-9964.1 0.771038 << 0.00001 Square_1/_Enthalpy_of_vaporization_at_the_boiling_point__kJ/mole
223906 ----- 1/_Enthalpy_of_vaporization_at_the_boiling_point__kJ/mole__
359 260.272
-7814.9 0.769263 << 0.00001 Hydrogen_bond_number 9.20856 ----- 1/_Enthalpy_of_vaporization_at_the_boiling_point__kJ/mole__
359 207.119
-7419.51 0.768355 << 0.00001 surface_tension 1.39539 ----- ==================================================
Transformed variable models for
Flash_Point__C_ Variable df Intercept Coefficient r squared probability x transform:Flash_Point__C_ 1/_Enthalpy_of_vaporization_at_the_boiling_point__kJ/mole__
303 248.543
-7919.01 0.831664 << 0.00001 sqrt(Log_P) 6.12759 ----- x transform:sqrt(Flash_Point__C_) 1/_Enthalpy_of_vaporization_at_the_boiling_point__kJ/mole__
264 18.1367
-437.486 0.805329 << 0.00001 sqrt(Log_P) -0.16568 ----- x transform:Flash_Point__C_ 1/_Enthalpy_of_vaporization_at_the_boiling_point__kJ/mole__
317 220.523
-7183.72 0.8041 << 0.00001 sqrt(CNDO_LogP) 13.1627 ----- x transform:Flash_Point__C_ 1/_Enthalpy_of_vaporization_at_the_boiling_point__kJ/mole__
329 304.009
-8328.6 0.792738 << 0.00001 sqrt(LUMO) -73.1153 ----- x transform:Flash_Point__C_ 1/_Enthalpy_of_vaporization_at_the_boiling_point__kJ/mole__
359
367.166 -5176.06 0.790236 << 0.00001 1/ln(hydrophilic_surface_area)
[+10.3280102539062]
-456.501 ----- ==================================================
Cross products, divisions and subtractions
of 2 variables for Flash_Point__C_ Variable df Intercept Coefficient r squared probability 1/_Enthalpy_of_vaporization_at_the_boiling_point__kJ/mole__
303 168.192
-7165.99 0.845318 << 0.00001 sqrt(Log_P)-surface_tension_in_water^2
-0.104649 ----- 1/_Enthalpy_of_vaporization_at_the_boiling_point__kJ/mole__
303 210.892
-6852.07 0.841105 << 0.00001 sqrt(Log_P)*surface_tension_in_water^2 0.0183651 ----- 1/_Enthalpy_of_vaporization_at_the_boiling_point__kJ/mole__
303 153.8
-7871.62 0.839776 << 0.00001 surface_tension_in_water-sqrt(Log_P) 4.21706 ----- 1/_Enthalpy_of_vaporization_at_the_boiling_point__kJ/mole__
292 274.532
-8036.35 0.838247 << 0.00001 sqrt(Log_P)*1/connectivity_3 -18.7291 ----- 1/_Enthalpy_of_vaporization_at_the_boiling_point__kJ/mole__
303 258.244
-8332.88 0.838074 << 0.00001 water_content_70%_RH__moles_-sqrt(Log_P)
-9.86567 ----- ==================================================
Two variable models for Flash_Point__C_ Variable df Intercept Coefficient r squared probability 1/_Enthalpy_of_vaporization_at_the_boiling_point__kJ/mole__
303 37.8682
-6105.12 0.856495 << 0.00001 water_solubility -4.28045 surface_tension_in_water-sqrt(Log_P) 7.20953 ----- 1/_Enthalpy_of_vaporization_at_the_boiling_point__kJ/mole__
303 -43.2173
-5512.44 0.856362 << 0.00001 Log_P 4.47645 surface_tension_in_water-sqrt(Log_P) 9.30119 ----- 1/_Enthalpy_of_vaporization_at_the_boiling_point__kJ/mole__
303 127.598
-6009.68 0.854661 << 0.00001 water_solubility -2.88661 sqrt(Log_P)-surface_tension_in_water^2
-0.123239 ----- 1/_Enthalpy_of_vaporization_at_the_boiling_point__kJ/mole__
303 17.2268
-4808.2 0.854414 << 0.00001 ln(1/_connectivity_1_) [+0.01] -34.3467 surface_tension_in_water-sqrt(Log_P) 4.67267 ----- 1/_Enthalpy_of_vaporization_at_the_boiling_point__kJ/mole__
303 208.448
-7299.08 0.854263 << 0.00001 percent_hydrophilic_surface 0.632755 water_content_70%_RH__moles_-sqrt(Log_P)
-16.5484 |
|
Figure 33.
Print-out for Regress with All
Combinations routine for the next iteration of the flash point model with
1/enthalpy of vaporization included in all models. The next step would be to graph the best
models and run them through the detailed multiple regression analysis with
PRESS. Then include another variable
and reiterate and so on until it is impossible to add another variable with
statistical significance (p for every variable must be <0.05). The two variable model containing 1/enthalpy of
vaporization and square root of log P (r square = 0.83) looks like a possible
improvement over the two variable model previously investigated. |
Alternatively instead of the Regress with All Combinations tool we could use the Stepwise routine to quickly look for
models up to 10 or even 20 variables (the Regress al Combinations tool is much
more thorough, but takes several iterations of model checking and building the
Included variable list - i.e. it will take some time to build a large
model).
Always ask the question does the model make good
chemical sense. Be particularly
suspicious when a new independent variable is added that is highly correlated
to another independent variable, as it is likely that is explaining one or two
inaccurately measured data points.
Examine the plot of residuals to make sure they look random. If not you may have the wrong variables in the
model or need a transformation of the dependent variable or one or more
independent variables. As you add
variables and the model grows in complexity it may be harder to judge by
intuition whether the model makes sense.
Modeling a subset of the data
The flash point data for low molecular weight
molecules (and low connectivity index 1)
when graphed appeared better correlated to enthalpy of vaporization than
the whole data set. You can do a
separate analysis for just this subset.
To subset the data, go to the Edit menu and select Subset by text or numeric value.
Choose to subset by connectivity_1 and choose the maximum value of 4.5
to subset by. Rerun the analysis. What you will find is that you will not have
to transform the enthalpy of vaporization values, that the model r squares are
much improved, but that the model standard deviation is significantly lower -
so the models obtained will be more accurate than the ones for the whole data
set when predicting flash point values of solvent sized molecules. You can restore the original data set by
going to the Edit menu and selecting Restore
subsetted data.
Restricting the size further by subsetting the data
with maximum molecular weight of 100 and allowing values from the literature in
the calculations gives these "best" models when Regressed with All Combinations:
Flash point
(C) = -53.3981 + 0.68443*Literature_Boiling_Point_C
N=227, r squared = 0.850624, p<< 0.00001
And
Flash point
(C) = -52.4996 -12.0857*CIM_7 +
0.760654* Literature_Boiling_Point_C
N = 209, r squared =
0.913709, p<< 0.00001
Analyze
Data Manually, Part 3 - PLS
In parts 1 and 2 we looked at linear least squares
regression techniques in generating a model for flash point. In this section we will look at use of
partial least squares regression to model the same flash point data as in parts
1 and 2.
Partial least squares (PLS) analysis is an alternative
to ordinary multiple regression analysis that has advantages if:
a) your causal (x, independent) variables are
inter-correlated. For instance here is
the correlation matrix for one of the models presented in Part 2 (correlation
matrix obtained from Principle Components Regression routine).
Correlation matrix for regression variables
Flash_Point 1/_Enthalpy sqrt(log P)
Flash_Point__C_ 1. -0.90519 0.73105
1/_Enthalpy_of_vaporization -0.90519 1. -0.78007
square_root(Log_P) 0.73105 -0.78007 1.
As you can see, enthalpy of vaporization and
the square root of log P are highly intercorrelated.
b) you have a high ratio of causal (x, independent)
variables to observations (multiple regression works best if there are no less
than 4 observations per causal (independent) variable).
Multiple regression may still be used in the above
cases, but answers may be “biased”. The
PLS technique, popularized in the late 1980s, attempts to address these
problems.
Application to the flash point model:
Like the other routines, this analysis uses the
industrial.mdb database that comes with the program. Open this database and go to the Analysis
menu of the database window. Select
Partial Least Squares Regression from the menu.
Select flash point from the list of fields and restrict the number of
principle components to 25. After quite
a while (these calculations take time) you will be presented with some
preliminary cross validation results, and the program will ask you to choose
the number of principle components to use.
|
|
Figure 34. It
is good for models have the cross-validation ration (PRESS/sum of squares of
Y) of <0.5. In fact none of the
models here meet that criterion. The
"sweet spots" (minimized cross-validation ratios) are for the 1
component and 4 component models. We
will examine the 4 component models more thoroughly below. |
|
|
Figure 35.
Normally we would not be interested in any of the higher principle
component models shown here because of the very high cross-validation
ratios. In fact each additional
component is explaining the variance for one or fewer data points in these
models. However, we will ignore this
for purposes of demonstration and look at the 18 component model. |
|
|
Figure 36. After
you select the number of components the program will more quickly calculate
the final model. Then it will offer
the user a chance to look at the models graphically. You should always look at the plots of
observed vs. predicted values and observed or predicted values versus
residuals. |
|
|
Figure 37.
The plot of the four component models of flash point (observed versus
predicted). There is one obvious
out-lier that should be examined and explained or removed. |
|
|
Figure 38.
The 18 component model of flash point.
There are three data points clumped that may be out-liers. If they are similar types of molecules that
occur no where else in the data set, they can be removed as long as in the
instructions to those using the model a warning is given that the model does
not apply to this type of molecule. |
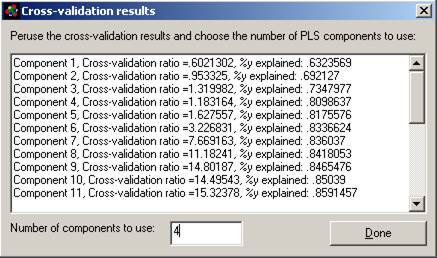
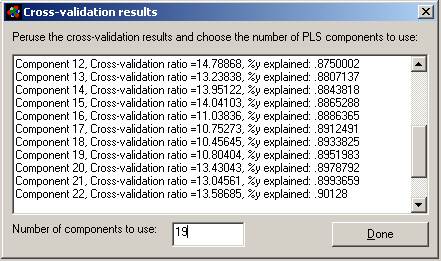
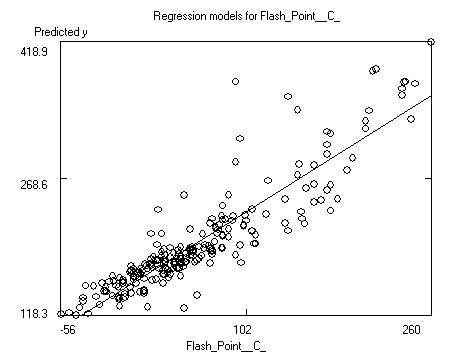
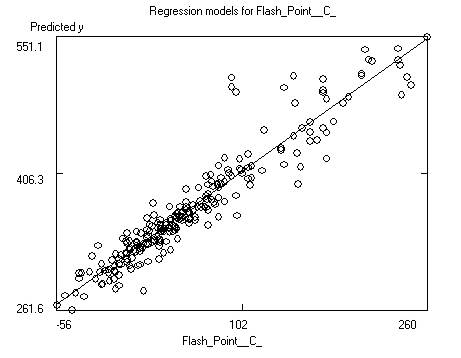
|
PLS component 1 : X variance explained, this component: 0.558255525962759 accumulated:
0.558255525962759 Y variance explained, this component: 0.632356946005208 accumulated:
0.632356946005208 PLS component 2 : X variance explained, this component: 0.210855353598904 accumulated:
0.769110879561663 Y variance explained, this component: 5.97700779226291E-02 accumulated:
0.692127023927837 PLS component 3 : X variance explained, this component: 0.104635496368257 accumulated: 0.87374637592992
Y variance explained, this component: 4.26706340999879E-02 accumulated:
0.734797658027825 PLS component 4 : X variance explained, this component: 2.86296954681797E-02 accumulated:
0.9023760713981 Y variance explained, this component: 7.50660083082563E-02 accumulated:
0.809863666336081 ---------------------------------- PLS Model Regression Coefficients: ---------------------------------- Y = Flash_Point__C_ --
Intercept: -137.9093 --
molecular_weight: 0.0275296 --
molecular_volume: -8.78747E-04 --
molecular_length: -1.13613E-03 --
molecular_width: 1.87833E-03 --
molecular_depth: -1.01165E-03 --
surface_area: -1.458387E-03 -- Log_P:
-2.320458E-03 -- HLB: 6.647635E-03 -- Hansen_polarity: 3.332011E-03 --
Hansen_hydrogen_bonding: 1.253919E-02 --
H_bond_acceptor: 2.942572E-04 --
H_bond_donor: 3.558267E-04 --
dipole_moment: -4.520623E-04 --
percent_hydrophilic_surface:
3.796799E-02 --
mean_water_of_hydration: 8.610905E-04 --
boiling_point: 0.1456266 --
connectivity_0: -5.591225E-03 --
connectivity_1: 1.293165E-03 --
connectivity_2: 1.053856E-03 --
connectivity_3: 1.748378E-03 --
valence_0: -7.125869E-03 --
valence_1: -3.463916E-04 --
valence_2: -4.808458E-04 --
valence_3: 2.566669E-06 -- kappa_2:
-6.578427E-04 --
water_solubility: 2.305892E-03 --
connectivity_index_4: 1.428772E-03 --
valence_index_4: 4.468389E-05 --
dipole_moment_from_CNDO: -1.795975E-02 --
H_bond_acceptor_from_CNDO: -7.366852E-05 --
H_bond_donor_from_CNDO: 1.266422E-04 --
HOMO: 1.2663E-05 -- LUMO:
-8.185258E-05 --
CNDO_LogP: 1.281364E-04 --
LogP_-_Crippen: -1.456506E-03 --
surface_tension: 1.707584E-02 -- hydrophilic_surface_area: 3.162643E-03 --
surface_tension_in_water: 1.941342E-03
--
Critical_Temperature__K_: 0.1099477 --
Critical_pressure__bar_: 1.194533E-02 --
Critical_volume__cm^3/mole_: -3.065991E-02 --
Normal_boiling_point__K_: 0.1845125 --
Normal_freezing_point__K_: 0.157135 --
Enthalpy_of_formation_ideal_gas_at_298_K__kJ/mole_: 4.978785E-02 --
Gibbs_energy_of_formation_ideal_gas_unit_fugacity_at_298_K__kJ: -7.055692E-02
--
Enthalpy_of_vaporization_at_the_boiling_point__kJ/mole_: 2.585548E-02 --
Enthalpy_of_fusion__kJ/mole_:
2.627288E-03 --
Liquid_viscosity__N_s/m^2_:
1.769484E-03 --
Heat_capacity_ideal_gas__J_mole_K_: -5.038143E-02 --
Effective_number_of_torsional_bonds: -2.295547E-03 -- Hydrogen_bond_number: 6.985848E-06 --
entropy_of_boiling__J/K_mole_:
9.00867E-03 --
Heat_capacity_change_at_boiling__J/K_mole_:
4.82065E-03 --
CIM_1: 1.034556E-04 --
CIM_2: 2.440436E-04 --
CIM_3: 1.272485E-04 --
CIM_4: 3.572252E-05 -- CIM_5: 1.473604E-04 --
CIM_6: 1.433094E-04 --
CIM_7: 2.177646E-04 --
CIM_8: 2.6544E-04 --
1/_Enthalpy_of_vaporization_at_the_boiling_point_kJ/mole__: -1.769506E-05 --
square_root(Log_P): 1.338915E-04 |
|
Figure 39. Part
of the print-out from the partial least squared routine. The "corrected" r squared values
and the model coefficients are listed in this part of the print-out. Examination of the model coefficients
(multiply by the average size of the values) can lead to elimination of some
of the fields from subsequent analyses (eliminate the fields that contribute
little to the predicted flash point value). |
Substructure and Similarity Searches
Molecular Modeling Pro allows several kinds of
structural searching routines in the database window.
1. Load a database.
2. Draw a substructure in the Drawing window (e.g. a
substructure is a part of a molecule)
3. In the Edit Menu of the Database window select
substructure search. If you have not run
this before, the program will create a new database of substructural keys for
every molecule in the database (i.e. the number of carbons, the number of
double bonds, the number of amide groups etc.).
Note that for very large databases, this will take some time. Incidentally, you can load and use this
database in Molecular Modeling Pro Plus (load an ssf file from the File
menu). The program will identify all the
structures in the database which contain the substructure drawn in the drawing
window. A list of matching structures
will be displayed. Click on the names to
4. Alternatively you can find an exact match of the
molecule drawn in the Drawing window by selecting “Exact Match for Structure”
from the Edit menu of the database window.
After the search is complete, the database record that matches will be
highlighted in the spreadsheet.
The program also looks for the most similar molecules
in a database to the molecule drawn in the drawing window. This is done using the Compare menu in the
database window.
1. Load a database
2. Draw a molecule in the drawing window (or get a
structure from the database).
3. There are then the following options for doing a
similarity search
a. Find the molecules which are closest in all the
calculated properties in the database
b. Find the molecules which are closest in the 3-D
solubility parameters (dispersion, polarity and hydrogen bonding). This might be useful for solvent or
surfactant replacement in a formulation.
This routine is not affected by molecular conformation.
c. Find the molecules which are closest in the Rule of
Five bioavailability parameters (log P, molecular weight, hydrogen bond
acceptor and donor, polar surface area).
This might be useful for determining which molecules in a database of
analogs might have the closest property to a known drug for uptake. This
routine is not affected by molecular conformation.
d. Find the molecules which most closely match in 3-D
structure – looks at size, shape and distribution of hydrogen bonding
atoms. This search might be used as a
screening tool to see if any molecules in a large database should be tested for
biological or other activity similar to the molecule drawn in the Drawing
Window. This may require the creation of
a large database of 3-D properties as a preliminary step, so can take some
time. This routine is affected by 3-D
conformation. Similar molecules that are
stored with different shapes and dimensions due to being in different conformations
will not be found to be similar.
Molecular orientation, on the other hand, does not affect the outcome of
this search.
e. Find the molecules which have the most sub-structural
features in common. This should find the
closest analogs of the molecule drawn in the Drawing Window. This search is independent of conformation
and uses the sub-structural keys database.
If this database is not set up, you must do so by running the
Substructure Search routine from the Edit menu (can take some time for a very
large database).
f.
The final option,
“Find Matching Combinations of Molecules” is the most flexible structural
search done by MMP+. You will be asked
to select the properties to search under (MMP+ must be able to calculate these
fields). You will also be given the
opportunity to exclude molecules in the database from the search. This routine will not only print out the most
similar molecules, but will also look at combinations of two molecules. The combinations are calculated at 10%
intervals by simply multiplying the fractions times the property values and
adding them together. This routine may
have some utility in formulation research where the investigator may be
interested in replacing a formulation ingredient with a mixture of
ingredients. The more similar the
ingredients in the mixture are, the more likely the result is likely to work
for this use.
Other
programs
Molecular Modeling Plus has
built-in ties to several other programs.
1. Support of MDL molfiles
over the clipboard. You can move
molfiles from one program to another over the clipboard between Molecular Modeling
Pro Plus and Chemsite, Accord, Chemical Inventory System and other programs
which support this file transfer in the public part of the clipboard.
2. There are also built-in
access through the Tools, Geometry and Display menus to Chemsite. There are tie-ins to Microsoft Excel in the
File menu of the database window.
3. The companion database, ChemicaElectrica, was created using Molecular Modeling Pro Plus and
can be viewed, editing and added to using MMP+.
This database contains over 5500 pharmaceutical and industrial chemicals
with information on use, trade names, and over 110 fields of chemical
properties.